In Article 120-123, the MDR establishes its transitional provisions, including the transition periods. However, the descriptions of these transitional provisions and transition periods are worded in a very complex manner. As a result, manufacturers are at risk of misunderstanding them and therefore not complying with regulatory requirements or incurring unnecessary costs.
A flow chart in chapter 2 of this article summarizes the regulatory requirements and provides clarity. It is also available as a free download.
This article considers the further postponement of the MDR transition periods adopted by the EU Parliament in February 2023 and the June 2024 amendment of Regulation 2024/1860 on the “gradual roll-out” of EUDAMED.
1. MDR transition periods: Introduction
The discussion regarding transition periods is usually limited to the question of how long a device can be placed on the market. As a result, the MDR has a number of transition periods.
Manufacturers need, for example, to differentiate between the following aspects with regard to transition periods:
- Placing on the market of legacy devices
- Making legacy devices available and putting them into service
- OEM-PLM setup
- Person Responsible for Regulatory Compliance
- Post-Market Surveillance
- Vigilance
- Quality System
- UDI
- Clinical Investigations
This article provides an overview of these transition periods.
2. MDR transition periods at a glance
a) Summary
| Aspect | Transition period | Comment |
|---|---|---|
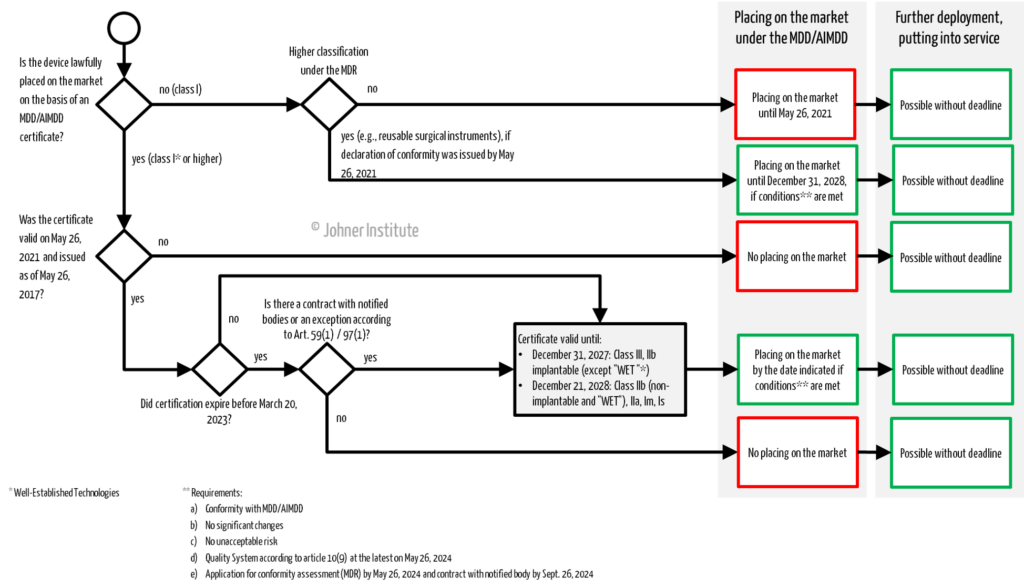
| Placing on the market of legacy devices | see flow chart (Fig. 1) | It is about the first making available |
| Making legacy devices available and putting them into service | — | The restrictions (“sell-off rule”) have been removed by the amending regulation. |
| OEM-PLM setup | The same transition periods apply to these devices. | The PLM must have the technical documentation to meet the MDR requirements for post-market surveillance and vigilance. |
| Person Responsible for Regulatory Compliance | — | Not required for manufacturers of legacy devices. See the detailed transition periods paragraph in the PRRC article (German). |
| Post-Market Surveillance | May 26, 2021 | With restrictions regarding EUDAMED; no restriction on monitoring and reporting (e.g., PSUR). |
| Vigilance | May 26, 2021 | With restrictions regarding the EUDAMED |
| Quality System | May 26, 2021 (MDR-devices) May 26, 2024 (legacy devices) | The amending regulation requires an MDR Article 10-compliant quality system for manufacturers of legacy devices. |
| UDI | see tab. below | |
| Clinical Investigations | May 26, 2021 | Initiated inspections may continue, but new reporting requirements apply. |
Article 10 of the MDR does not apply to devices that benefit from the transition period. Explicitly excluded from this are the abovementioned requirements for post-market surveillance and vigilance, among others.
The summary can be found in this fact sheet for download.
In August 2023, the EU also published a flow chart on its website.
Download md_devices-art120_flowchart.
b) Placing on the market, making available, and putting into service
How long manufacturers can still place their existing devices under the old directives (MDD/AIMDD) on the market, make them available or put them (or have them put) into service depends, among other things, on the class of the device and the validity of any certificates.
If you still have questions about switching to MDR, feel free to benefit from the Johner Institute’s free micro-consulting service.
c) EUDAMED obligations (e.g., registration)
| First 6 months after publication of the declaration that the respective EUDAMED module is fully functional | From 6 months after publication | Up to 12 months after publication | Up to 18 months after publication | |
| MDR devices and legacy devices | No requirements | Registration of stakeholders, registration for clinical investigations, vigilance, PMS, registration of new MDR devices and certificates by notified bodies, and upload of SSCPs (see Article 123(3)) | Completing the registration of legacy and MDR devices that were placed on the market before the 6 months after publication | Completion of the registration of certificates by notified bodies and upload of SSCPs for devices that were placed on the market before the 6 months after publication |
According to this publication in the Federal Gazette, special transitional provisions apply to economic operators based in Germany. Accordingly, manufacturers and their Authorized Representatives must register directly in EUDAMED and not in the national BfArM database, DMIDS, from May 26, 2021. The same applies to importers of MDR-compliant medical devices.
d) Transition periods for the UDI
The date from which the UDI must be applied depends on the class of the device:
| Class | Time |
|---|---|
| I | May 26, 2025 |
| IIa | May 26, 2023 |
| IIb | May 26, 2023 |
| III and implantable devices | May 26, 2021 |
For reusable devices where the UDI has to be placed on the device itself, the MDR grants two additional years.
However, these transition periods regarding UDI do not apply to legacy devices, i.e., devices “which are compliant with the Medical Device Directives (MDD and AIMDD) and placed on the market after the application date of the Regulations“. There are two other documents to consider here:
In the FAQ, the EU writes:
In order to facilitate the transition to the new system, the new Regulations give manufacturers the possibility to place products on the market after the general application dates of the new Regulations (and until 26 May 2024 at the latest) by virtue of valid Directive certificates. These legacy devices are not subject to UDI obligations but they should be registered in the Eudamed database. Timelines for registration as described under question 6 also apply to these products. More information on the operational aspects of the registration of legacy devices is available at the MDCG 2019-5 guidance document.
FAQ of the EU
e) Transition period for the Person Responsible for Regulatory Compliance
If manufacturers do not place devices on the market under the MDR, but only legacy devices according to art 120 (3), Article 15 of the MDR does not yet apply under MDCG 2021-25. This means that these manufacturers do not yet have to designate a Person Responsible for Regulatory Compliance (PRRC).
Until now, we and also many others had interpreted this differently. The reason was that the obligations for post-market surveillance and vigilance must be fulfilled by all manufacturers from May 26, 2021. The PRRC is responsible for an effective post-market surveillance and vigilance process. The MDCG 2021-25 guidance also considers this aspect but concludes that article 15 is not yet applicable to manufacturers of legacy devices.
All manufacturers must implement effective processes for post-market surveillance and vigilance in all cases, only monitoring by the PRRC is not yet mandatory for manufacturers of legacy devices.
Details can also be found in the article on the PRRC (German) in the section “transition periods”.
3. The MDR Articles 120 to 123
a) MDR, Article 120 Paragraph 2
Original text
Article 120 is entitled “Transitional provisions”. It also mentions the transition periods. The second paragraph is particularly relevant:
Certificates issued by notified bodies in accordance with Directives 90/385/EEC and 93/42/EEC as of May 25, 2017, which were still valid on May 26, 2021, and which have not been subsequently withdrawn, shall remain valid after the end of the period indicated on the certificate until the date specified in paragraph 3a of this Article for the relevant risk class of devices. Certificates issued by notified bodies in accordance with these Directives as of May 25, 2017, still valid on May 26, 2021, and expired before March 20, 2023 shall be considered valid until the dates specified in paragraph 3a of this Article only if one of the following conditions is met:
MDR Article 120 (2)
a) Before the expiry of the certificate, the manufacturer and a notified body have signed a written agreement in accordance with the second subparagraph of section 4.3 of Annex VII to this Regulation on the conformity assessment of the device covered by the expired certificate or of a device intended to replace that device;
b) a competent authority of a Member State has granted an exemption from the applicable conformity assessment procedure in accordance with Article 59 (1) of this Regulation or has requested the manufacturer to carry out the applicable conformity assessment procedure in accordance with Article 97 (1) of this Regulation
Interpretation of this text
If the manufacturer has a certificate issued before May 26, 2017, under the guidelines, it remains valid until the specified date. This excludes certificates issued under Annex IV of the Directives (“batch certification”), which were valid until May 27, 2022, at the latest.
If, on the other hand, the notified bodies’ certificate was issued on or after May 26, 2017, and was still valid on May 26, 2021, these will remain valid until the stated deadlines (see Paragraph 3a). However, if these certificates expired before the amendment regulation came into force, i.e., before March 20, 2023, the extended validity will only apply if:
- the manufacturer has entered into a contract with a notified body for MDR certification BEFORE the expiration of the certificate (in accordance with Annex VII, Section 4.3, Subparagraph 2 of the MDR), and the contract has been signed
- OR the manufacturer has been granted special authorization by the national competent authority in accordance with Article 59 (1) MDR
- OR further placing on the market has been permitted by the competent authority even without a valid certificate, subject to a deadline (Article 97 (1) MDR).
b) MDR, Article 120 Paragraphs 3a and b
Original text
(3) By way of derogation from Article 5 and provided that the conditions referred to in paragraph 3c are met, the devices referred to in paragraphs 3a and 3b may be placed on the market or put into service until the dates referred to in those paragraphs.
MDR Article 120 (3)
(3a) Devices for which a certificate valid by virtue of paragraph 2 of this Article has been issued in accordance with Directive 90/385/EEC or Directive 93/42/EEC may be placed on the market or put into service until the following dates:
(a) December 31, 2027, for all class III devices and class IIb implantable devices, except sutures, staples, dental fillings, braces, dental crowns, screws, wedges, dental or bone plates, wires, pins, clamps, and connectors;
(b) December 31, 2028, for class IIb devices other than those referred to in subparagraph (a) of this paragraph, class IIa devices, and class I devices placed on the market in a sterile condition or with a measuring function.
(3b) Devices for which the conformity assessment procedure under Directive 93/42/EEC did not require the involvement of a notified body, for which the declaration of conformity was drawn up before May 26, 2021, and for which the conformity assessment procedure under this Regulation requires the involvement of a notified body, may be placed on the market or put into service until December 31, 2028.
Interpretation of this text
The third paragraph of Article 120 (a) and (b) can be interpreted as follows:
Legacy devices with a valid directive certificate may be placed on the market up to the specified time limits, depending on the risk class.
- Class III, class IIb implantable devices (except those devices with “well-established-technology”): December 31, 2027
- Other class IIb, IIa, Is, and Im devices: December 31, 2028
- Class I devices under the Directives that are higher classified under the MDR and thus require the involvement of a notified body in the conformity assessment procedure (e.g., reusable surgical instruments, many software products): December 31, 2028
However, these extended transition periods only apply under certain conditions. These are mentioned in Paragraph 3c.
c) MDR Article 120 Paragraphs 3c and d
Original text
(3c) Devices referred to in Paragraphs 3a and 3b of this Article may be placed on the market or put into service only until the dates referred to in those Paragraphs if the following conditions are met:
a) The devices continue to comply with Directive 90/385/EEC or Directive 93/42/EEC, as the case may be;
b) there are no significant changes in the design and intended use;
c) the devices do not pose an unacceptable risk to the health or safety of patients, users, or other persons or to other aspects of public health protection;
d) the manufacturer has established a quality management system in accordance with Article 10, Paragraph 9, no later than May 26, 2024; and
e) the manufacturer or the authorized representative has, no later than May 26, 2024, submitted a formal application to a notified body in accordance with the first Subparagraph of Section 4.3 of Annex VII for conformity assessment for a device referred to in Paragraph 3a or 3b or for a device intended to replace that device, and the notified body and the manufacturer have signed a written agreement in accordance with the second Subparagraph of Section 4.3 of Annex VII no later than September 26, 2024.
(3d) By way of derogation from Paragraph 3 of this Article, the requirements of this Regulation on post-market surveillance, market surveillance, vigilance, and registration of economic operators and devices shall apply to devices referred to in Paragraphs 3a and 3b instead of the corresponding requirements of Directives 90/385/EEC and 93/42/EEC.
Interpretation of this text
The third paragraph of Article 120 (c) describes the conditions that must be met for the application of the extended transition periods.
Requirement 1
The device continues to comply with the requirements of the MDD or AIMDD. This sounds logical and should also be ensured by the monitoring of the notified bodies and authorities.
Requirement 2
The manufacturer has not made any significant changes to the device or intended purpose. What exactly is meant by this is explained by the MDCG in the document MDCG 2020-03.
The question of how significant changes may be so that they do not have to be considered “significant” is a particular cause for discussion. Please refer to the article on design changes.
Requirement 3
That the devices do not pose an unacceptable risk to safety and health should be ensured by compliance with the directives. Nevertheless, this is a monitoring task of the notified bodies and surveillance authorities. This is also made clear by the MDCG in guidance document MDCG 2022-04. This contains recommendations on the surveillance activities of legacy devices during the transition periods. It states:
“In cases where the audit activities reveal a major non-conformity, which may present an unacceptable risk to the health or safety of patients, users or other persons, the notified body needs to take action, i.e. suspend, restrict or withdraw the certificate, and inform the relevant competent authority.“
Requirement 4
The requirements for a complete quality system, according to Article 10, Paragraph 9, have been supplemented by the amendment Regulation. Thus, it is no longer sufficient to implement only the processes for post-market surveillance and vigilance, according to the MDR. The latter must already be implemented in an MDR-compliant manner since May 26, 2021 (see 3 (d)).
Requirement 5
This condition should also not be underestimated and should not be neglected. Manufacturers must have submitted an application for MDR certification to a notified body by May 26, 2024. It is unclear whether the application must only have been submitted by then or whether it must also have been positively examined by the notified bodies. This leaves room for interpretation and differences of opinion. We currently assume that this date only concerns the application but not the inspection by the notified bodies. The possibility of applying for MDR certification not for the actual legacy product but for a “replacing device” is interesting. However, it is unclear what the requirements are for this. For example, must the replacing device be equivalent, or can it be based on a different technology? Unfortunately, this also leaves room for interpretation. The MDCG should provide clarity on this as soon as possible.
In addition to the application, a signed contract with a notified body for MDR certification must be available by September 26, 2024.
d) MDR Article 120 Paragraph 3e
Original text
(3e) Without prejudice to Chapter IV and Paragraph 1 of this Article, the notified body that issued the certificate referred to in Paragraph 3a of this Article shall remain responsible for the appropriate monitoring of all applicable requirements for the devices it has certified unless the manufacturer has agreed with a notified body whose designation has been made in accordance with Article 42 that it will perform such monitoring.
MDR Article 120 (3)
No later than September 26, 2024, the notified body that signed the written agreement referred to in Paragraph 3c (e) of this Article shall be responsible for monitoring the devices covered by the written agreement. Where the written agreement concerns a device intended to replace a device for which a certificate has been issued in accordance with Directive 90/385/EEC or Directive 93/42/EEC, the monitoring shall be carried out in relation to the replaced device.
The arrangements for the transfer of monitoring from the notified body which issued the certificate to the notified body whose designation has been made in accordance with Article 42 shall be clearly set out in an agreement between the manufacturer and the notified body whose designation has been made in accordance with Article 42 and, where practicable, the notified body which issued the certificate. The notified body whose designation has been made in accordance with Article 42 shall not be responsible for conformity assessment activities carried out by the notified body that issued the certificate.
Interpretation of this text
Paragraph 3e regulates the surveillance activities and responsibilities of the notified bodies. As a rule, the notified body that issued the directive certificate is responsible for further monitoring within the transition periods. However, manufacturers may change their notified bodies, which will then take over the surveillance activities. This may be necessary, for example, if the current notified body has not (yet) been designated under the MDR. By September 26, 2024, at the latest, a notified body designated under the MDR must then have monitoring responsibility.
e) MDR Article 120 Paragraph 3f
Original text
(3f) By way of derogation from Article 5, custom-made implantable devices of class III may be placed on the market or put into service until May 26, 2026, without a certificate issued by a notified body in accordance with the conformity assessment procedure referred to in the second Subparagraph of Article 52 (8), provided that the manufacturer or the authorized representative has submitted a formal application for conformity assessment to a notified body in accordance with Section 4.3, first Subparagraph, of Annex VII no later than September 26, 2024, and that the notified body and the manufacturer have concluded a written agreement in accordance with Section 4.3, first Subparagraph, of Annex VII no later than September 26, 2024. May 2024 at the latest to a notified body, and the notified body and the manufacturer have signed a written agreement in accordance with the second Subparagraph of Section 4.3 of Annex VII.
MDR Article 120 (3)
Interpretation of this text
Paragraph 3(f) was added by the amending regulation. The MDR now also grants an extended transitional period until May 26, 2026 for Class III implantable custom-made devices. The conditions are less stringent in this case. The MDR “only” requires a formal application for conformity assessment by May 26, 2024 and the signed contract by September 26, 2024.
f) MDR Article 120 Paragraph 4
Original text
The fourth paragraph is comparatively easy to understand:
(4) Devices lawfully placed on the market pursuant to Directives 90/385/EEC and 93/42/EEC prior to 26 May 2021, and devices lawfully placed on the market from 26 May 2021 pursuant to paragraphs 3, 3a, 3b and 3f of this Article, may continue to be made available on the market or put into service.
MDR Article 120 (4)
Interpretation of the text
This means that there is no sell-off period anymore. This provision is intended to ensure that “safe and important medical devices that have already been placed on the market continue to be available to healthcare systems and the patients who rely on them.”
A distinction must be made between the terms “making available” and “placing on the market.” You can find a definition of the terms in our article on placing on the market.
g) MDR Article 122 – dash 1 and 2
Article 122 causes many readers to shake their heads. Who is supposed to understand this on first reading?
“-Articles 8 and 10, Article 10b(1), points (b) and (c), and Article 10b(2) and (3) of Directive 90/385/EEC, Article 10, Article 14a(1), points (c) and (d), Article 14a(2) and (3) and Article 15 of Directive 93/42/EEC, and the obligations relating to vigilance and clinical investigations provided for in the corresponding Annexes to those Directives, which are repealed, as applicable, with effect from the date referred to in Article 123(3), point (d), of this Regulation in respect of the application of the obligations and requirements that relate to the electronic systems referred to in Article 33(2), points (e) and (f), respectively, of this Regulation;
MDR Article 122, dash 1 and 2
-Article 10a, Article 10b(1), point (a), and Article 11(5) of Directive 90/385/EEC, Article 14(1) and (2), Article 14a(1), points (a) and (b), and Article 16(5) of Directive 93/42/EEC, and the obligations relating to registration of devices and economic operators, and to certificate notifications, provided for in the corresponding Annexes to those Directives, which are repealed, as applicable, with effect from the date referred to in Article 123(3), point (d), of this Regulation in respect of the application of the obligations and requirements that relate to the electronic systems referred to in Article 33(2), points (a) to (d), respectively, of this Regulation;“
Interpretation of this text
The requirements of the old directives mentioned in the two dashes concern the following obligations:
- reporting of incidents
- registration of economic operators
- device registration
- certificates of notified bodies
- reporting/approval of clinical investigations
- reporting and monitoring procedures by authorities
Article 122 thus states that the obligations mentioned in the old directives remain valid until the corresponding modules in EUDAMED are functional, and this has been announced by the Commission (see Article 123). Thus, for example, registrations of actors and devices must continue to be carried out nationally.
For those who want to understand Article 122 in more detail, here are some important references:
- AIMDD (90/385/EEC)
- Article 8: Information on incidents occurring after the device has been placed on the market
- Article 10a: Registration of the manufacturer
- Article 10b (1) (a): Certificates in the database
- Article 10b (1) (b) and (c): Data obtained in accordance with the vigilance procedure and data relating to clinical investigations
- Article 11(5): Certificates issued and withdrawn by notified bodies
- MDD (93/42/EEC)
- Article 10: Information on incidents occurring after the device has been placed on the market
- Article 14(1) and (2): Registration of persons responsible for placing devices on the market
- Article 14a (1) (a) and (b): European databank (manufacturer, authorized representative, certificates)
- Article 14a (1) (c) and (d): European databank (data from the vigilance procedure, data on clinical investigations)
- Article 15: clinical investigations
- Article 16 (5): certificates issued and withdrawn by notified bodies
h) MDR Article 123 (“Entry into force and date of application”)
Article 123 lists further deadlines that affect economic operators, e.g.:
- Paragraph 3(d): In the context of EUDAMED, this lists all the obligations that must be complied with no later than six months after the date on which the respective EUDAMED module is declared fully functional, and this is announced in the Official Journal of the EU.
- Paragraph 3(e): Manufacturers have 12 months to register legacy and MDR devices placed on the market before the six months after publication referred to in (d).
- Paragraph 3(ea) and (eb): For the devices referred to in (e), notified bodies have 18 months to store certificates and upload SSCPs.
- Paragraph 3, point (ec): If manufacturers are required to submit a report (e.g., serious incident, trend, field safety corrective action) and the corresponding devices have not yet been registered in EUDAMED, this must be done immediately. This only applies to MDR devices, not to legacy devices.
- Paragraph 3(f): The UDI carrier must be affixed to implantable devices and class III devices from May 26, 2021, to class IIa and IIb devices from May 26, 2023, and to class I devices from May 26, 2025. This exemption does not apply to the UDI assignment.
- Paragraph 3(g): For reusable devices that require direct marking, the deadlines mentioned in (f) are extended by two years each.
4. Other requirements
The following documents are regulatory relevant and should be considered:
- Application of transitional provisions concerning validity of certificates issued in accordance to Directives 90/385/EEC and 93/42/EEC
- MDCG 2019-15 GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES
- MDCG 2022-4 Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR with regard to devices covered by certificates according to the MDD or the AIMDD
5. Conclusion and summary
The pressure was great, and the complaints about the negative effects of the MDR could not be ignored by the EU Commission. It, therefore, postponed the transition periods several times. However, it has only partially remedied the problems as a result. Many harms have already occurred:
- Manufacturers have given up
- Devices have been taken off the market
- New devices are approved in the U.S. first
- The development of new devices has been abandoned or put on hold
Nevertheless, the extension of the transition periods was an important step. However, the specifications, which have been revised several times, are now difficult to keep track of and still leave questions unanswered.
Change history:
- 2024-10-11: Revision due to the amendment to the regulation on the gradual roll-out of EUDAMED
- 2024-06-07: Chapter 3c): Reference and citation from the MDCG 2022-4 updated
- 2023-08-25: In chapter 2a) Flowchart of the EU added
- 2023-03-20: Date of entry into force of the amending regulation added
- 2023-02-24: Article completely revised after adoption by parliament
- 2023-02-16: Note that proposal is before parliament added
- 2023-01-12: Added outlook and link to commenting possibility
- 2023-01-10: Note on planned changes to transition periods added
- 2022-03-04: Note on MDCG 2022-4 added
- 2021-06-14: Link to revised article on PRRC added, addressing transition periods in this regard
- 2021-05-17: Added note on UDI requirements for legacy devices in chapter 4d)

Liebes Team,
vielen Dank für diesen Beitrag. Eine Sache bleibt weiterhin ungeklärt: “Wie ist die Situation zu bewerten, wenn ein Hersteller die Zulassung eines Produkts gemäß den Anforderungen der MDR geplant und alle erforderlichen Anträge konform bei der benannten Stelle eingereicht hat, sich jedoch im Verlauf der genehmigten Übergangsfrist entscheidet, das Produkt vom Markt zu nehmen, weil der Zulassungsprozess wirtschaftlich nicht mehr tragbar ist? Gelten in diesem Fall weiterhin die Übergangsregelungen, oder verlieren diese ihre Gültigkeit mit der Entscheidung zur Marktrücknahme und was wären die weiteren Folgen?
Ich könnte mir vorstellen, dass vielen Herstellern erst im Nachgang klar wird, wie hoch der tatsächliche Aufwand und die Kosten für eine Produktakte gemäß den Anforderungen der MDR sind.
Guten Tag,
entscheidend in diesem Fall ist, ob die Voraussetzungen für die Übergangsfristen nach MDR weiterhin erfüllt sind. Falls der Antrag bei der benannten Stelle zurückgezogen oder der Vertrag beendet wird, ist die Voraussetzung nach 120(3c) (e) MDR nicht mehr erfüllt, d.h. Sie könnten nicht mehr von den Übergangsfristen profitieren.
Freundliche Grüße
Luca Salvatore
Liebes Team,
gibt es auch Umsetzungsfristen nach Ausstellung eines MDR-Zertifikats?
Wie lange darf ich nach Ausstellung des MDR-Zertifikats noch unter MDD weiter produzieren?