Because the quality of medical device sterilization is crucial to the safety of these medical devices, the selection and validation of the sterilization process are regularly subject to audits and inspections.
This article provides a quick overview of the various sterilization processes and regulatory requirements for medical devices and offers best practices for successfully passing inspections by authorities and notified bodies.
1. Sterilization: The context
a. Definition
In medicine and hygiene, sterilization is defined as a validated process for rendering medical devices, instruments, or materials completely free of germs. This process kills or removes all forms of microbial life, including bacteria, viruses, fungi, and their spores.
Medical devices are considered germ-free if the probability of a viable microorganism being present on the product is less than 10−6. Normatively (DIN EN 556), this means that less than one microorganism per million units (SAL value: Sterility Assurance Level 10−6) may be present. The medical device is then considered sterile.
The sterilization of reusable medical devices is a special case of medical device reprocessing.
b. Examples
- Medical devices that come into contact with broken skin (e.g., sterile wound dressings) or are even implanted are usually marketed in a sterile state by the manufacturer.
- In the case of reusable products such as surgical instruments, however, the responsibility for sterilization lies with the operator (hospitals, practices). Both manufacturers and operators can use external sterilization service providers for this purpose.
- On the other hand, products that only come into contact with intact skin (e.g., walking aids) or have no patient contact (e.g., software) do not require sterilization.
In some cases, both manufacturers and operators use sterilization service providers.
2. Sterilization procedures
a. Overview
Several methods are available for sterilizing medical devices, which are more or less suitable depending on the application.
| Method | Principle | Special features | Examples |
| Steam sterilization (Moist heat) | Saturated steam at high temperature and pressure | Very robust, established process Not suitable for materials that are sensitive to heat or moisture | Metal surgical instruments, textiles (surgical drapes/clothing), reusable medical devices |
| Ethylene oxide sterilization (EO) | Gas sterilization at low temperatures | Very good penetration of porous materials Residues (EO, ECH, ethylene glycol) must be monitored Long process and ventilation times | Catheters and tubes, products made of plastics and elastomers, textiles with elastane/polymers, complex assemblies with cavities, medical devices with electronics |
| Gamma sterilization | Ionizing radiation (cobalt-60) | Industrial process, high throughput rates May cause material aging or changes as well as discoloration Depends on the material | Disposable syringes, cannulas, surgical gloves, implants, packaged disposable products, suture material |
| Electron beam sterilization (E-beam) | Accelerated electrons | Shorter process times than gamma Lower penetration depth → restrictions for thick/complex products | Single-use items with low material thickness, syringes, tubing, flat plastic products, packaged disposable products |
| Low-temperature plasma (e.g., hydrogen peroxide plasma) | Reactive plasma species at low temperature | Short cycles Limited material and geometry compatibility Primarily for reprocessing, less for industrial series production | Endoscopes (limited, depending on the system), temperature-sensitive instruments, optical components |
When chemical sterilization is mentioned, this usually refers to sterilization with ethylene oxide (EO). Other chemical processes are special cases in the context of medical devices (e.g., peroxide).
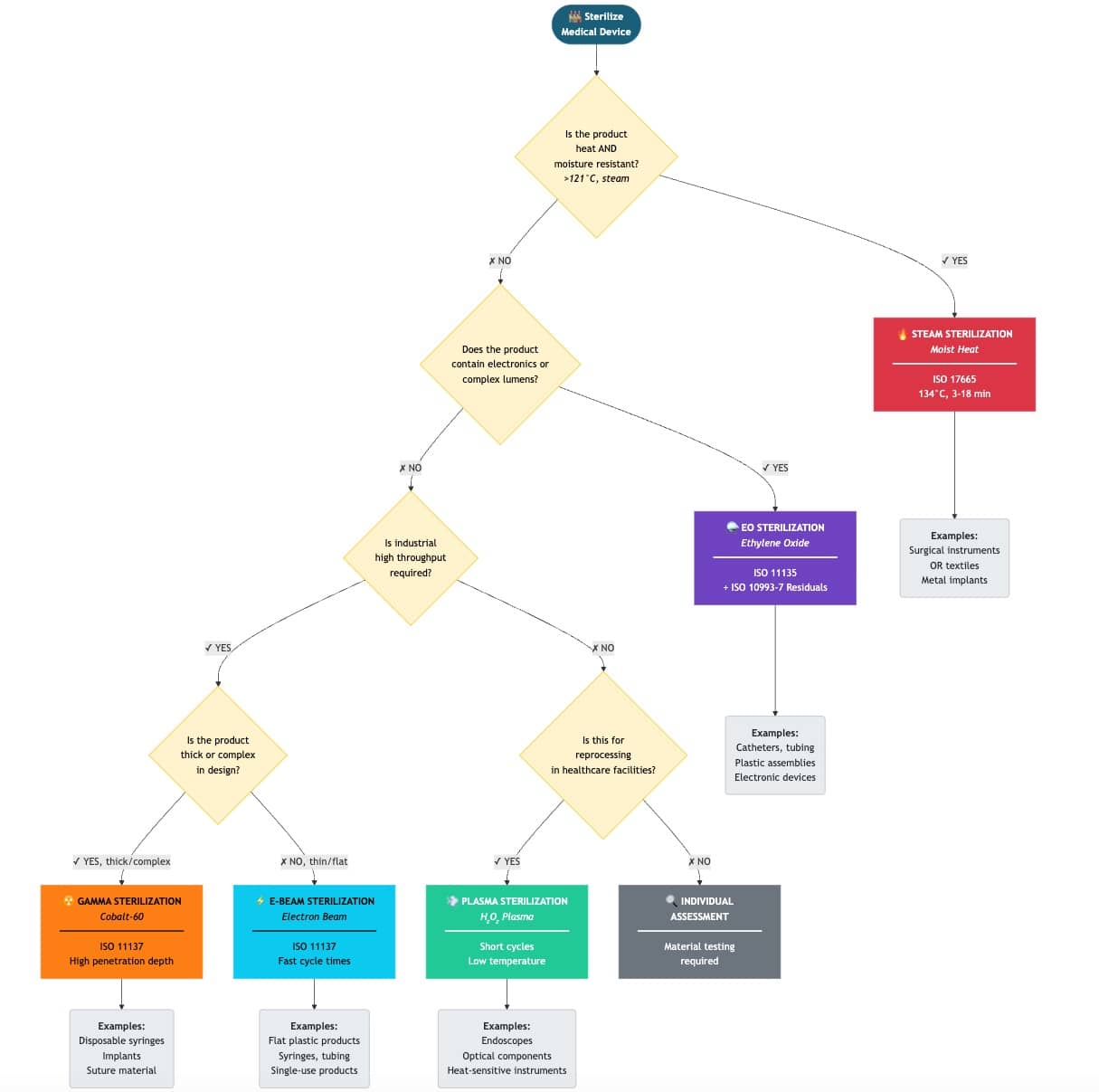
b. Selection
The selection of a sterilization method depends on the attributes mentioned above. Manufacturers can document their selection in the form of a decision diagram, as illustrated in Fig. 1.
3. Regulatory requirements for sterilization
a. MDR requirements
The MDR requirements on sterilization can be found in Annex I, Chapter II, Section 11.1:
“Products placed on the market in a sterile condition must be manufactured and sterilized using appropriate and validated processes.”
b. Normative requirements
Manufacturers also demonstrate compliance with these regulatory requirements by observing relevant (harmonized) standards.
| Standard | Content |
| ISO 11135 | EO sterilization |
| ISO 17665 | Moist heat/steam sterilization |
| ISO 11137-1/-2/-3 | Radiation sterilization |
| ISO 11138 series | Biological indicators |
| ISO 11140 series | Chemical indicators |
| EN 556-1 | Requirements for “STERILE” labeling |
| ISO 10993-7 | EO residues |
ISO 13485 also specifies requirements for process validation.
c. FDA requirements
Requirements for manufacturers
510(k) submissions for sterile products must include detailed descriptions of the sterilization process, packaging, and validation. Changes to the sterilization process often require a new evaluation or a new 510(k) submission.
The FDA also emphasizes the need to consider environmental impacts when selecting a sterilization process, while patient safety remains the top priority.
The FDA provides information on the most important requirements for the sterilization of medical devices. It also refers to the above-mentioned standards.
Requirements for contract sterilizers
Companies that offer contract sterilization services are required to register their facilities with the FDA annually.
4. Tips for legally compliant sterilization of medical devices
a. Take a structured approach
As with any process, a structured approach is essential when sterilizing medical devices:
- Precisely define the intended purpose and intended use of the medical device. This includes:
- Product life
- Qualification of users (including those responsible for sterilization)
- Number of reprocessings
- Requirements for the sterilized medical device
- Select a suitable sterilization method and determine parameters
- Consideration of material stability with the selected method
- Define a suitable packaging system
- Validate sterilization process
- Create a validation plan
- Establish validation infrastructure
- Perform validation
- Create validation report
b. Ensure competencies
Manufacturers should pursue the selection and validation of the sterilization process using an interdisciplinary approach. Depending on availability, it is advisable to involve experts from fields such as materials science, biocompatibility, sterilization, and product development in order to comprehensively cover technical, biological, and regulatory requirements.
c. Consider parametric release
Parametric release makes it possible to dispense with costly and time-consuming batch-dependent sterility tests. This requires validated process parameters that ensure reliable control and reproducibility of the sterilization process. Robust data on the basic microbiological load can play a decisive role.
d. Observe the special conditions of the respective sterilization method
Each sterilization method has specific requirements in terms of materials, packaging, and process control. For example, EO sterilization requires precise control of humidity, temperature, and gas concentration, as well as a subsequent desorption phase to minimize toxic residues. In radiation sterilization, on the other hand, material compatibility and dose distribution are paramount, while steam sterilization requires, in particular, the thermostability of the product and packaging.
5. FAQ
a. Which sterilization method is suitable for plastics?
EO and radiation sterilization are primarily suitable for plastics, as both methods operate at low temperatures and are gentle on thermally sensitive materials. Steam sterilization can only be used for thermally stable plastics.
The decisive factors for the choice of process are material compatibility, product geometry, and regulatory requirements, which are taken into account during product development and process validation.
b. How often must sterilization validation be repeated?
A complete revalidation is not required by regulation at fixed intervals, but is required in the event of significant changes to the product, packaging, or process.
The relevant standards (ISO 11135, ISO 11137, ISO 17665) also require at least one routine monitoring per year to continuously confirm the validated status (existing products).
Immediately after sterilization validation (new product/new process), quarterly reviews are initially required for at least one year to take seasonal influences into account. Once stable and robust process performance has been demonstrated, the frequency can then be reduced to semi-annual and later to at least annual monitoring.
In addition, ISO 13485 and the MDR require regular assessment of process capability as part of the quality management system.
c. What are typical errors that are noticed in audits?
- Incomplete or outdated validation documentation: Protocols, reports, or master plans do not correspond to the current process status.
- Inadequate routine monitoring: Missing or delayed dose audits, bioburden reviews, or process parameter controls
- Poor change control management: Changes to the product, packaging, or loading are not systematically evaluated against the validated state.
- Missing or incomplete risk assessment: No traceable link between risk management (ISO 14971) and validation activities
- Incorrect or insufficient justifications for worst-case selection or formation of product families
- Deficiencies in supplier qualification: Inadequate contractual agreements and lack of regular audits of external sterilization service providers
d. What should you look for when selecting contract sterilizers?
The following criteria are helpful when selecting contract sterilizers:
- Certification, completeness of the QM system
- Integration into your own QM system
- Experience with the required sterilization process
- Experience with the product classes and their special features (e.g., geometries, materials)
- Proximity, logistical connections
- Throughput times
- Reporting, transparency (e.g., statistics), process records
- Size, history, financial stability
- Price
5. Conclusion and summary
The procedures for sterilizing medical devices are mostly proven. Nevertheless, authorities and notified bodies often encounter difficulties. These are mainly caused by medical device manufacturers failing to comply with regulatory requirements, not taking into account the specifics of their products, or violating best practices.
This article has shown how manufacturers can avoid these difficulties by taking a structured approach and ensuring competence.
The experts at the Johner Institute support medical device manufacturers, for example, by reviewing existing validations. In this way, they ensure that residues from EO sterilization are checked and toxicologically evaluated in accordance with ISO 10993-7.
To this end, the experts also consider the validation process from a biocompatibility perspective and ensure that the evidence required by regulations is complete and compliant and that patient safety is guaranteed.