The De Novo request, which the FDA also refers to as the “De Novo program” and the “De Novo submission process”, is one of the approval processes for medical devices in the United States.
As the name “De Novo” suggests, manufacturers can use this process for novel products.
In other words, manufacturers should submit a De Novo application for devices for which there is no comparable legally marketed device available (a “predicate device”), but which the FDA would likely classify as class I or class II.
1. Background and introduction
a) The problem
Manufacturers are not allowed to use the 510(k) clearance procedure for new types of devices because there is no comparable predicate device. The FDA has not yet defined a classification for these new type of devices, which is why they would fall into class III by default (see FD&C Act, 513(f)(1)).
Manufacturers of novel devices or class III devices must undergo a PMA approval process. However, this “Premarket Approval” (PMA) is just as expensive as it is labor and time-intensive. This would be overwhelming for manufacturers of non-critical devices.
b) Solution
The FDA has launched the De Novo classification request to limit these disproportionate efforts. This is a process in which the FDA
- decides about the classification,
- assigns a new classification regulation and product code for the new device type and
- defines the associated “controls”. Examples of controls are special labeling requirements, design, or production controls, i.e., aspects of a quality management system. You can find an overview of the controls here.
A device granted as “De Novo” can serve as a “predicate device” for similar devices.
2. Procedure of the De Novo classification request
a) Variants
There are two ways to begin the process (see Fig. 1):
- “classic” De Novo
The manufacturer submits a 510(k) application (Premarket Notification, PMN). The FDA will issue an NSE letter, because the device is not “substantially equivalent” (NSE) to the predicate device. In their NSE decision, the FDA may express its opinion that the device could be a De Novo candidate. Regardless of this assessment, manufacturers may submit a De Novo application refering to the NSE decision.. - “direct De Novo”
Alternatively, manufacturers are also allowed to submit a De Novo classification request directly. This is recommended if the manufacturer believe there is no suitable predicate device, and the FDA will classify the device into class I or II.
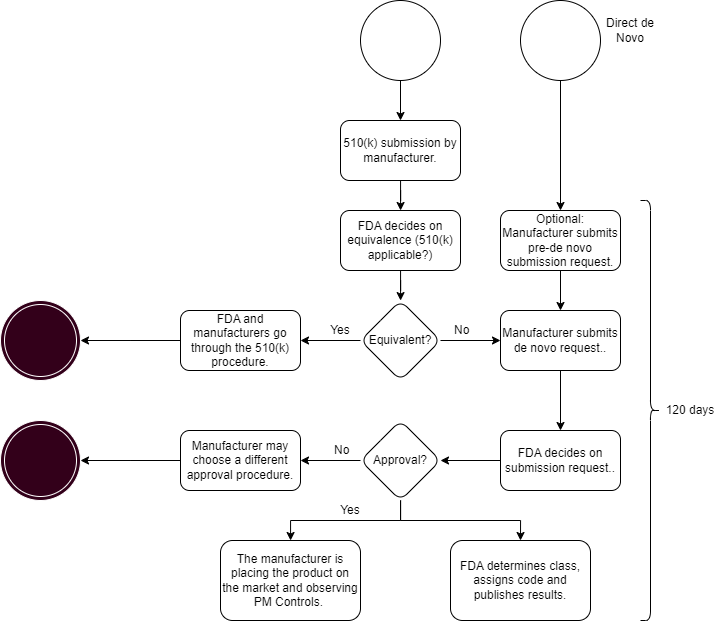
Fig. 1: Manufacturers have two options for submitting a De Novo application (click to enlarge)
In the second case, manufacturers may request a pre-submission meeting with FDA before the actual De Novo submission.
Read more about pre-submission requests here.
b) Review by the regulatory authority and timing
Formal review
The formal Refuse-to-Accept review will become obsolete with the obligation to use the eSTAR submission format starting October 1, 2025
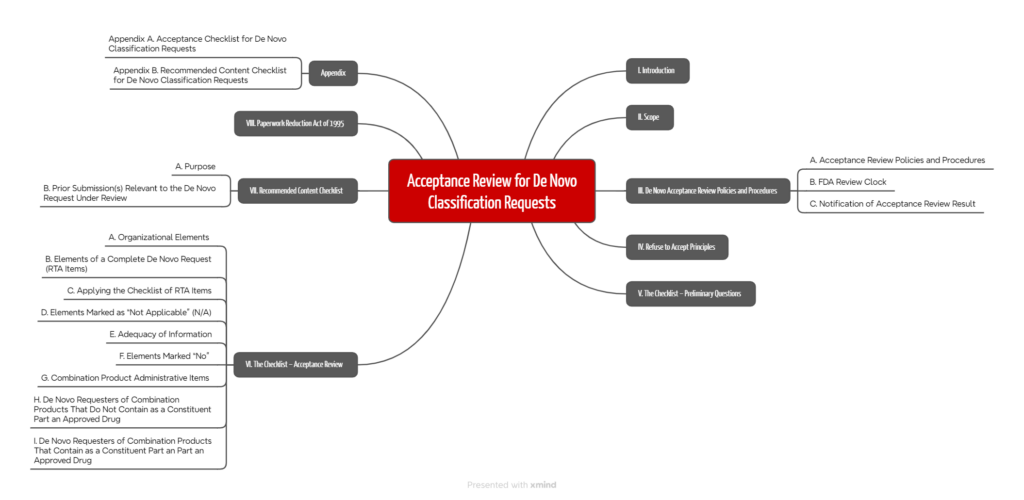
First, the FDA reviews the application using an “acceptance checklist” that can be found in Annex A of the Guidance Document “Acceptance Review for De Novo Classification Requests”. The agency intends to decide within 15 days whether to accept the application at all.
The FDA first assesses the application based on formal decision criteria. This includes determining whether
- the manufacturer has paid the fee,
- the device qualifies as a medical device,
- the application is addressed to the correct FDA center, and whether
- A De Novo request is appropriate for the device, e.g., if there is a predicate device, the request will not be granted.
If the application passes these initial steps, the FDA continues with the Annex A checklist and evaluates:
- Organizational aspects: Is the FDA able to navigate through the submitted application?
- Completeness: Have required contents incl. evidence been submitted for each section? r E.g., for software, the FDA would check whether the “documentation level” has been determined and whether the documentation is consistent with the guidance document “Content for the Premarket Submissions for Device Software Functions”.
- Combination products: For device-drug combination products, further documents are required. There is also the requirement that the drug has already been approved.
Substantive review
The FDA uses the same guidance document for its substantive review, applying the checklist in Annex B. The checklist includes questions on the following aspects:
- Description of the device, including its accessories, labeling for patients and users, etc.
- Justification of why the De Novo request is appropriate
- Identification of alternative procedures, technologies, devices
- Summary of preclinical data such as tests and clinical data from clinical studies
- List of standards applied (in particular “consensus standards”)
The time between submission of the application and the authority’s decision should not exceed 120 days, in case FDA does not request additional information
c) Submission contents
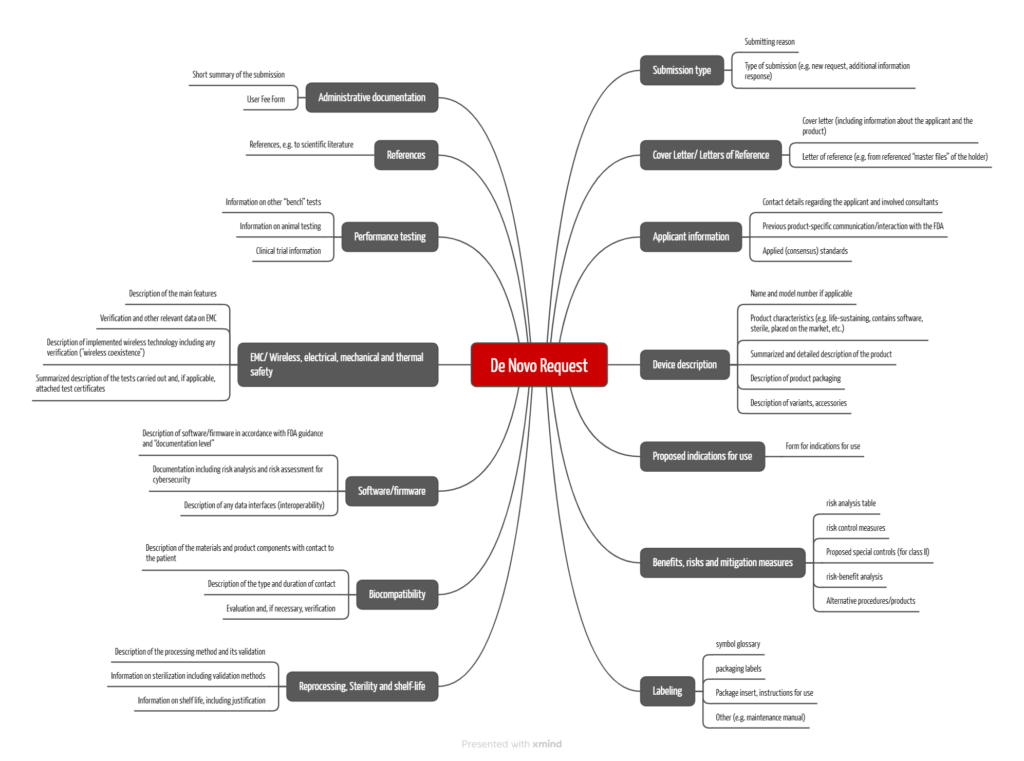
The FDA requires similar information for both variants mentioned under a). The FDA has already developed a structure for manufacturers to submit this information. For applications submitted on or after October 1, 2025, the submission will be in eSTAR format. The FDA describes the structure and content in the Electronic Submission Template for Medical Device De Novo Requests guidance document. This requires, among other things:
- Administrative information (manufacturer/applicant, product name, type of submission, etc.)
- Reference to any communication with the FDA and FDA history (e.g., IDEs, pre-submissions) before submission
- Applied Recognized Consensus Standards
- Product name and detailed description of the product, including intended use or indications for use
- Proposal for classification with justification
- Brief description of the benefits, the risk analysis and description of the risk controlmeasures
- Justification of why the device is safe and achieves the intended use using scientific evidence (risk-benefit discussion). Typically, clinical data relating to the deviceis required for this.
- For devices that are likely to fall into class II, a proposal for adequate “special controls” to minimize risks.
- “Labeling,” incl. instructions for use
- Information on reprocessing and its validation
- Information on sterilization and its validation
- Information on the “shelf life”
- Evaluation of the biocompatibility
- Particular information and evidence on software/firmware and cybersecurity, as well as interoperability (if applicable)
- Test results (e.g., laboratory tests, software tests, animal testing, clinical investigations)
- References (e.g., to scientific articles)
- Responses to FDA requests for additional information
As described above, the FDA requires the application to be submitted in eSTAR format with a predefined chapter structure (see Fig. 3).
3. Regulatory background
The Food, Drug & Cosmetic Act describes the De Novo classification process in Section 513(f) (2) of Title 21. It also refers to the procedure as “Evaluation of Automatic Class III Designation”.
This article makes it clear that this procedure is only permitted for devicesthat have not yet been classified.
4. Costs for a De Novo procedure
A De Novo request is not cheap (see “price list” ): InFDA fiscal year 2025, the FDA is charging over USD 162,000. The FDA reduces the fee to just around USD 40,000 for small businesses.
5. Conclusion
The De Novo classification request is recommended for novel and less critical devices. Careful preparation is the key to success here as well. Manufacturers must clearly demonstrate
- why there is no comparable predicate device on the market,
- the justification for the proposed classification and “controls”,
- what risks exist, and
- how these are controlled so that the benefits outweigh the risks.
A clear intended use, a detailed description of the device, and sufficient evidence are a prerequisite for a successful De Novo request.
The Johner Institute helps manufacturers quickly and efficiently:
- Deciding whether to submit a De Novo request directly
- Reviewing whether the application is complete to avoid unnecessary loss of time or even rejection
- Deciding whether a pre-submission application or a 510(k) submission is the better strategy
- Completing missing information and improving applications
- Participating in pre-submission meetings
- Take over communication with the FDA
This enables manufacturers to minimize approval risks and thus avoid unnecessary costs and delays, bringing their devices to market quickly and safely.
Are you planning to have your device approved in the USA? Then get in touch with us.
Change history:
- 2024-10-11: Revision due to the eSTAR format required from 2025
