The Medical Device Regulation (MDR) has significantly increased the requirements for distributors. Learn to understand these requirements to avoid multi-year custodial sentences threatened in the event of infringement.
This article also considers extensive guidance issued by the Irish regulator.
1. Distributor: definition & delimitation
a) Definition
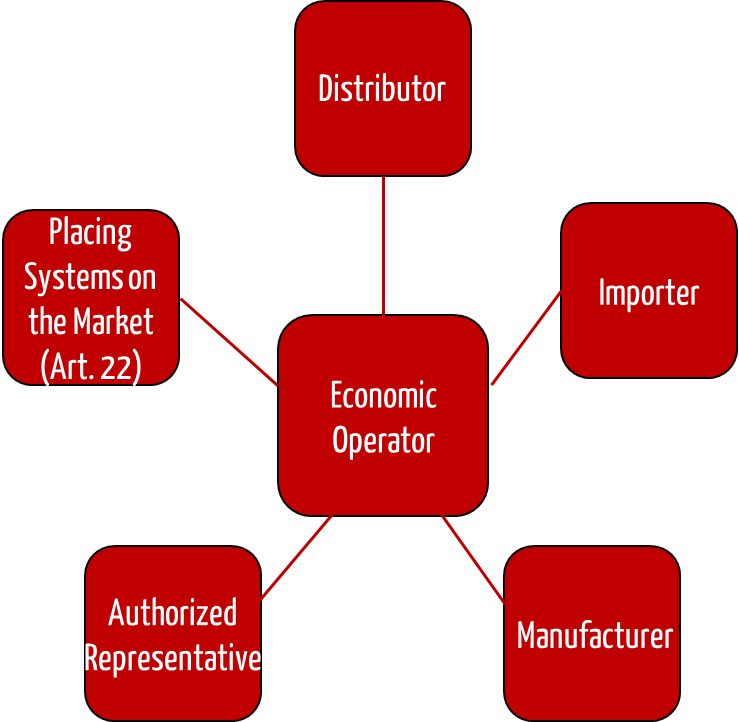
The Medical Device Regulation (MDR) has introduced many roles, including that of economic operators. In addition to manufacturers, these economic operators include authorized representatives in the EU, importers, and distributors.
“distributor” means any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting into service;
Source: MDR, Article 2
Distributors are persons or organizations selling medical devices, whether to end users or intermediate distributors.
b) Typical activities in the distribution of medical devices
The typical activities of a distributor include:
- Purchase of products from manufacturers or other (intermediate) distributors
- Marketing and sale of products to end-customers or other distributors
- Storage and transport of products
- Possibly attaching own labels (Be careful here! More on this later.)
- Instruction of users
- Support in installation and putting into service
- Answering user questions
- Handling customer complaints, providing feedback to manufacturer
Some distributors also deal with the service and repair of products or organize these.
c) Delimitation of the roles of distributor and importer
If a distributor purchases the products from a manufacturer or another distributor that is not based in the EU, this distributor also takes on the role of the importer. The MDR defines importers as follows:
“importer” means any natural or legal person established within the Union that places a device from a third country on the Union market;
Source: MDR, Article 2
The MDR imposes additional requirements on importers.
d) Delimitation of the roles distributor and manufacturer
Many distributors may sell the products under their own name and not name the actual manufacturer. Thus the distributor becomes the manufacturer. The Medical Device Regulation defines manufacturers as follows:
“manufacturer” means a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trade mark;
Source: MDR, Article 2
With the MDR, the PLM-OEM constructs are also a thing of the past. For this, the MDR, under Article 2 (16) (2) offers distributor new options: It no longer sees the following activities as a product change that has an effect on the conformity of the product:
- Provision of information for a product. This also applies to translations.
- Changes to the external packaging
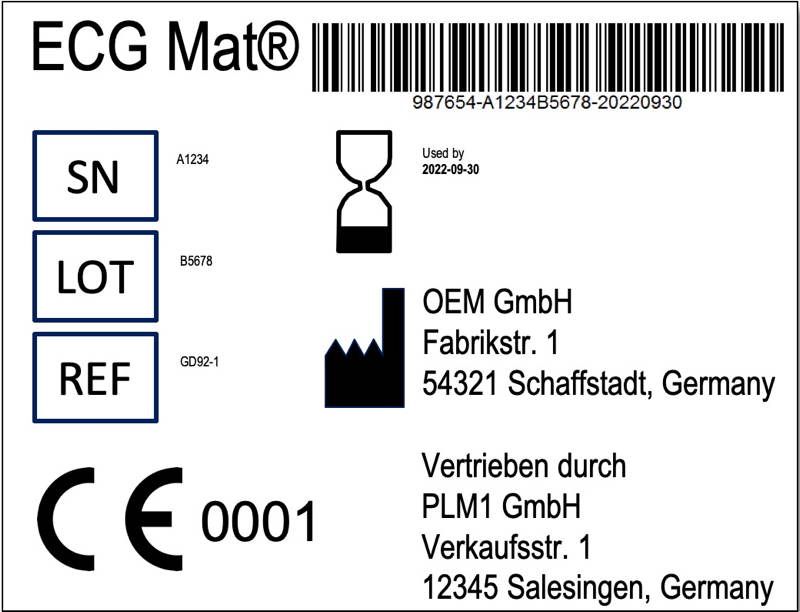
Of course, it would also be conceivable for the manufacturer to market the product in the distributor’s “design.” However, the manufacturer must always (also) be recognizable on the label. This is required by Article 16, Paragraph 3.
2. Obligations of distributors
Even before distributors sell medical devices, they must comply with many legal requirements.
a) Background and history
The requirements set for the distributor by the Medical Device Regulation MDR are derived from a superordinate framework for marketing products. This is also called the “Goods Package” and is based on:
- EU Directive 765/2008 “on the regulations for accreditation and market surveillance within the context of marketing of products”
- Resolution no. 768/2008/EU “on a common legal framework for the marketing of products”
b) Obligations of distributors as inspection entities in the supply chain (from the perspective of the MDR)
The idea of the MDR is that each economic operator ensures, as far as possible, that the economic operator, one step earlier in the supply chain, has achieved regulatory compliance.

The reviews required by the MDR for distributors include:
- Does the product bear a CE mark?
- Has a declaration of conformity been issued for the product?
- Has the importer listed its name and address on the product, packaging, or a document?
- Has the importer not covered the labels of the manufacturer with its own additional(!) labels?
- Has the manufacturer issued a UDI?
- Does the product appear to conform to the legal requirements?
If one of these conditions is not fulfilled, the distributor may not sell the product and must inform the manufacturer, importer, and the EU representatives.
Distributors may no longer blindly relay on the manufacturer and importers. They must exactly review whether the manufacturer has valid certificates, in particular in the transition period.
c) Obligations of distributors as inspection entities in the supply chain (from the perspective of the MPDG)
MPDG requirements
The MDR allows distributors to perform random testing in Article 14 Paragraph 2. They should be able to review whether the products actually have a CE mark, a declaration of conformity, and a UDI.
The German MPDG (Medical Device Implementation Act) appears to be more rigid: There is a threat of a custodial sentence in § 92 Para. 1 No. 3 in conjunction with § 13 MPDG if someone offers falsified products, stores them, or puts them into operation. This affects the distributors directly.
The prohibition in § 13 MPDG and the associated threat of punishment must be seen in the context of Art. 14 (2) Subparagraph 3 MDR: According to this, there is a ban on distribution and an additional obligation to notify the competent authority if a distributor “believes or has reason to believe” that a device is falsified and therefore non-compliant.
However, in our view, neither this nor § 92 (1) No. 3 and § 13 MPDG impose a regulatory obligation on a distributor to go beyond random inspections to check for possible counterfeits, i.e., to be suspicious by default and to determine a 100% inspection based on this. For the penalty provision, we believe it also depends on whether the unauthorized offering or stockpiling of actual counterfeit goods was negligent or even intentional. It thus becomes critical if a distributor shows further indications or suspicions of distributing counterfeits.
Possible conflicts arising from this
If the distributor discovers a falsified product and keeps it in the warehouse, he is liable. However, according to §13 MPDG, returning the product is also problematic, because then it falls “out of the scope of validity of this law.” This is then also punishable.
Then there is still the possibility of destroying it. But even in this case, he allegedly makes himself liable to prosecution, because he must “provide free samples of the product at the request of the authorities,” or, “if this is not practical, allow access to the product” (MDR Article 14(6)).
A reader who brought our attention to this conflict (thank you for that), writes:
“I’m curious about the assistance of the legislators about how these contradictions should be solved by a distributor, e.g., by Aldi, which offers medical devices once or twice a year (e.g., blood pressure monitors) within the framework of any special offers.”
d) Requirements for the activities of the distributors
The MDR obligates the distributors to additional activities:
- Storage and transportation pursuant to manufacturer specifications
- Collecting complaints and incident reports and forwarding them to manufacturers and possibly importers. This also applies to products for which the distributor itself has doubts about the conformity.
- Keeping a “register” of non-conforming products, recalls, and withdrawals
- Informing authorities (in Germany the BfArM according to Article 44 of the MPDG) about unsafe and falsified products and about corrective actions, and provide information and documentation upon request
With the MDR, distributors and importers become part of the post-market surveillance and reporting system. They must actively cooperate in this, which requires that they be able to trace the products. This is explicitly described in the MDR in Article 25 and by the EU in its “Factsheet for Authorized Representatives, Importers and Distributors.”
e) Registration of distributors
In Article 30, the MDR mentions the “Electronic System for the Registration of Economic Operators.” In it, it only obligates the manufacturers, authorized representatives, and distributors. The MDR leaves it to the member states to issue conditions for the registration of distributors.
And that’s just what member states such as Germany appear to be doing: In the Medical Device Implementation Act MPDG is stated:
(1) The Federal Ministry of Health is authorized…
9. to determine that distributors making products available on the German market shall notify the competent authority of this prior to commencing their activities and to regulate the content and form of the notification.
MPDG § 88
Thus a national ordinance is to be expected, that describes this obligation to register.
f) Special case quality management system
When distributors need a quality management system
As described above, manufacturers may repackage products and provide and translate the legally required information (Annex I, Section 23). However, this not only results in the obligation mentioned above to indicate the distributor’s name on the label (see Fig. 2). Rather, the distributor must have a quality management system.
This QM system must regulate all relevant actions:
- Repackaging of the product (and checking that this repackaging does not affect the conformity)
- Creation/provision of information (and checking that these fulfil the legal requirements)
- Translating the information
- Receive/demand(?) information about the manufacturer’s corrective actions
According to the Irish authority, the QM system does not have to be certified: “it is not a requirement that a quality system is officially accredited to any specific standard.” However, this contradicts the requirement of the MDR.
Within the same period of 28 days, the distributor or importer shall submit to the competent authority a certificate, issued by a notified body designated for the type of devices that are subject to activities mentioned in points (a) and (b) of paragraph 2, attesting that the quality management system of the distributer or importer complies with the requirements laid down in paragraph 3.
MDR Article 16, Paragraph 4
Which processes a quality management system should define
Whether this QM system must meet a specific standard and be audited to a particular standard appears unclear. According to the Irish authority, this QM system must be relatively comprehensive and also guide other activities such as:
- Personnel, Training
- Control documents and records
- Reception, storage, and provision of medical devices
- Handling returned products
- Handling falsified products
- Recall of products
- Contracted processes
- Transport
- Audits
- Supplier management
- Management review
- CAPA
- Waste management
- Internal and external audits
- Validation including computerized systems validation (CSV)
Note, that as a distributor,
- the authority requires that you implement a complete QM system comparable to ISO 13485.
- in the case of Article 16(4), you must certify to the authority that your QM system meets the requirements of this article.
Presumably, so that the authority can check the QM system, the MDR requires distributors to notify it 28 days before the product is made available. The MDR uses the word “notify” rather than “request approval.”
We support both manufacturers and distributors in quickly establishing ISO 13485-compliant quality management systems, thus ensuring compliance with legal requirements.
g) Obligations not (?) imposed on the distributors by the MDR
The distributors are not obligated to register the products. This is the task of the manufacturers or importers. The distributors also do not require a “Person Responsible for Regulatory Compliance.”
It is unclear whether the distributor is responsible for the storage and transport if the manufacturer delivers directly to the distributor’s customers. The question of whether the importer or the distributor is responsible for the transport from the importer to the distributor also leads to discussion.
h) Transition periods
The so-called sell-through provision of MDR Article 120(4) primarily concerns distributors. It is intended to limit the period during which AIMDD/MDD-compliant devices that have already been placed on the market (for the first time) may be made available, e.g., by a distributor (either before the date of application or through Article 120(3) MDR after the date of application).”
After May 27, 2025, these devices may no longer be made available/put into service (= end date). Such devices still in the commercial chain on this date – i.e., have not yet been made available to the end user (e.g., hospital) as a ready-to-use device – are no longer “tradable.”
Art. 120(4) MDR essentially addresses the “making available” on the market of AIMDD/MDD compliant devices after they have been placed on the market (for the first time), e.g., in the chain of commerce. It does not regulate the “initial) placing on the market” of these devices by the manufacturer.
Translated from NAKI UG1 (German)
Furthermore, the German National Working Group for the Implementation of the MDR/IVDR (NAKI) clarifies that “trade in second-hand devices should not be covered by the so-called “sell-through” provision (see recital 32). This means that once a device has been made available to the end user (e.g., hospital) as a ready-to-use device, the further availability of that device on the market is not within the scope of the MDR.”
The MDCG clarifies in MDCG 2021-25 that economic operators must also comply with their obligations under MDR for legacy devices (Article 120, valid MDD certificate). For each economic operator, the MDCG lists the relevant articles of the MDR that they must comply with. For distributors, these are:
- Article 14(2), last section: This is about the information obligations in the context of post-market surveillance.
- Article 14(4): Again, this is about the information obligations and cooperation in the corrective actions.
- Article 14(5): This also deals with the cooperation in the context of the post-market surveillance and the related measures.
- Article 14(6): This paragraph concerns the cooperation of manufacturers with the authorities.
i) Clarification by the CJEU
There is a CJEU ruling on distributor obligations (C-10/24). The CJEU has clarified which verification obligations “distributors” have under Article 14 of the MDR—and where the boundaries lie.
The case: A distributor sold dental compressors bearing the CE marking as “machinery”—not as medical devices. However, the BfArM classified these as Class IIa accessories. A competitor issued a cease-and-desist letter.
The three key takeaways from the CJEU: Consistency check yes, conformity assessment no: Distributors must verify, based on available documentation, whether the CE marking and DoC obviously comply with the MDR—but they must not repeat the manufacturer’s assessment.
- Consistency check: yes; conformity assessment: no: Distributors must verify, based on available documentation, whether the CE marking and DoC obviously comply with the MDR—but they must not repeat the manufacturer’s assessment.
- Classification is the manufacturer’s responsibility—but for classes requiring a Notified Body (NB), the four-digit NB number must be verified.
- Warnings trigger a verification obligation: The distributor must consult the manufacturer and may rely on the manufacturer’s assessment as long as it is not obviously untenable.
Classification is the manufacturer’s responsibility—but for classes requiring a Notified Body (NB), the four-digit NB number must be verified.
Warnings trigger a verification obligation: The distributor must consult the manufacturer and may rely on the manufacturer’s assessment as long as it is not obviously untenable
Coherence check yes, conformity assessment no: Distributors must verify, based on available documents, whether the CE marking and DoC obviously comply with the MDR—but they must not repeat the manufacturer’s assessment.
Classification is the manufacturer’s responsibility—but for classes requiring a Notified Body (NB), the four-digit NB number must be verified.
Warnings trigger a verification obligation: The distributor must consult the manufacturer and may rely on the manufacturer’s assessment as long as it is not obviously untenable.
3. Notes for manufacturers (regarding distributors)
a) Problem
Some manufacturers think that all of these regulatory requirements are the distributors’ problems, and not their own. But that’s not the case:
Distributors may even “relabel” products and “repackage” them, e.g., in their own design. They must only “notify” the manufacturer, not ask for approval.
This means that on the one hand the manufacturers can’t (just) inhibit these activities. On the other hand, they are obligated to perform post-market surveillance. This is difficult if distributors sell products in other countries unknown to the manufacturer with translated accompanying materials.
b) Post-market surveillance
Manufacturers should proactively search for information about their own products worldwide. This explicitly does not apply to the countries in which they know their products are sold. Automation such as the Post-Market Radar of the Johner Institute helps here.
It is essential that the manufacturers, together with their distributors, authorized representatives, and importers, exactly determine who is responsible for which activities (not only) within the post-market surveillance.
It goes without saying that post-market surveillance should also focus on falsified devices and unauthorized distributors.
c) Setting the hard line for distributors
Manufacturers should monitor their distributors just like the products themselves.
Distributors who “repackage” or “relabel” the devices must even provide the authority with a certificate from a notified body. This relieves the manufacturers of some of the work.
Nevertheless, manufacturers cannot rely on this blindly. They must know and monitor their distributors.
Sometimes, it is helpful to choose the intended use and the design of the products (e.g., plugs) so that certain markets are excluded. This enables manufacturers to somewhat curb “misuse” by unwanted distributors.
In addition, manufacturers should make quality assurance agreements with distributors. Such a QAA should, for example, regulate:
- Affected products or product groups
- Duration of validity of the agreement
- Contacts for both parties
- Form and deadline for feedback from the distributors to the manufacturer
- The right of the manufacturer to audit/inspect the distributor
- Obligation of the distributor to report to authorities
- Obligation of the distributor to trace products (“register”)
- If necessary, prohibition on sale to other distributors
- Obligation of the distributor to inform importers and EU representatives
- Keeping test samples available
- Obligation to provide accompanying materials
- Obligation to only work with a certified translation agency
d) Caution with fulfillment partners
Certain logistics partners offer services that go beyond those of a standard parcel service provider. These so-called “fulfillment centers” or (“fulfillment houses”) store products, package them upon request, take over invoicing, etc.
Then, one no longer speaks of “neutral service providers” but of distributors in the sense of the MDR.
Our tip: Use the specifications of the Blue Guide of the EU.
The MDR has substantially increased the requirements for distributors. That is understandable because only all parties involved in the supply chain can jointly understand where which products are and whether these products are compliant.
4. FAQ
a) Who is authorized to sell medical devices?
The law does not exclude any persons or organizations from selling medical devices. However, the law does place requirements on these sellers, who are called distributors. The distributor obligations were presented in this article.
However, medical device manufacturers can prohibit the trade in their products (with restrictions).
b) What license or authorization is required to trade with medical devices?
There is no official permission or authorization by state authorities. However, distributors must comply with legal requirements before they are allowed to start trading. These requirements may include:
- Quality management system (for certain distributors)
- Registration with authorities (e.g., in Germany; see above)
- Reviews of the product (e.g., information from the importer, UDI)
If a distributor is also an importer, there are additional requirements that must be met before trading can begin.
In addition, a manufacturer may impose requirements on distributors. For example, the distributor may be required to provide proof of competence or take a test with the manufacturer.
c) How do you bring a medical device to market?
The EU distinguishes between placing on the market, first placing on the market, and making available. Distributors are natural or legal persons who make products available on the market.
Please note the article on placing medical devices on the market, which also explains the distinction of making them available.
5. Conclusion and summary
Economic operators can only respond to non-conformities and thus increase patient safety by working together. The MDR thereby removes the possibility of finger-pointing.
These are important findings:
- The distributors should have a (nearly) complete quality management system. At least, that is the view of the Irish authority. Processes should be established not only for the feedback system.
If a distributor carries out activities in accordance with Article 16(2), he must have a QM system. There is no (current) requirement for certification. - The manufacturers should be aware that the obligations of distributors also affect them, at least indirectly.
- The Irish authority has published a guideline for distributors with further practical tips.
It is to be hoped that the MPDG will not lead to contradictions with the MDR and that the result of these legal tightening will be greater patient safety and not more bureaucracy.
Change history:
- 2026-06-11: section 2.i) added, which heeds the new EuGH judgment
- 2024-12-18: FAQ (chapter 4) added; subheadings added to 2.e) and 2.f); main headings shortened; some editorial improvements
- 2021-10-25: In section 2(h), requirements added by MDCG 2021-25 guideline.

Your analysis of the MDR’s impact on distributor requirements is both clear and insightful! What key strategies do you recommend for distributors to effectively implement a comprehensive quality management system while ensuring compliance and enhancing patient safety?
Dear Dennis,
thank you very much for your question.
Establishing the Quality Management System for distributors or importers carrying out any of the activities mentioned in points (a)and (b) of Article 16(2) of the MDR shall be based on the MDCG guidance document 2021-23. This document contains all relevant processes which must be implemented in that case.
Best regards,
Manuela