The clinical evaluation is not a document but a process covering the entire product life cycle. Manufacturers use it to review the safety, performance, and effectiveness of their devices. This process produces documents such as
- the Clinical Evaluation Plan and
- the Clinical Evaluation Report.
Content
This page provides you with a quick introduction to the topic and links to relevant articles and further help:
- Basics
- Regulatory requirements
- Further articles
- Support with clinical evaluation
1. Clinical evaluation: the basics
a) Definition and objectives of the clinical evaluation
The MDR defines the term as follows:
“‘clinical evaluation’ means a systematic and planned process to continuously generate, collect, analyse and assess the clinical data pertaining to a device in order to verify the safety and performance, including clinical benefits, of the device when used as intended by the manufacturer;”
MDR Article 2, Sentence 44
The MDR thus also determines the objective: to review the safety, performance, and clinical benefit. Clinical evaluation is, therefore, a prerequisite for placing medical devices on the market.
The clinical evaluation must be based on clinical data to obtain the necessary clinical evidence.
b) Clinical evaluation in the product life cycle
The activities involved in clinical evaluation are closely interlinked with other activities in the product life cycle, such as development (see Fig. 1).
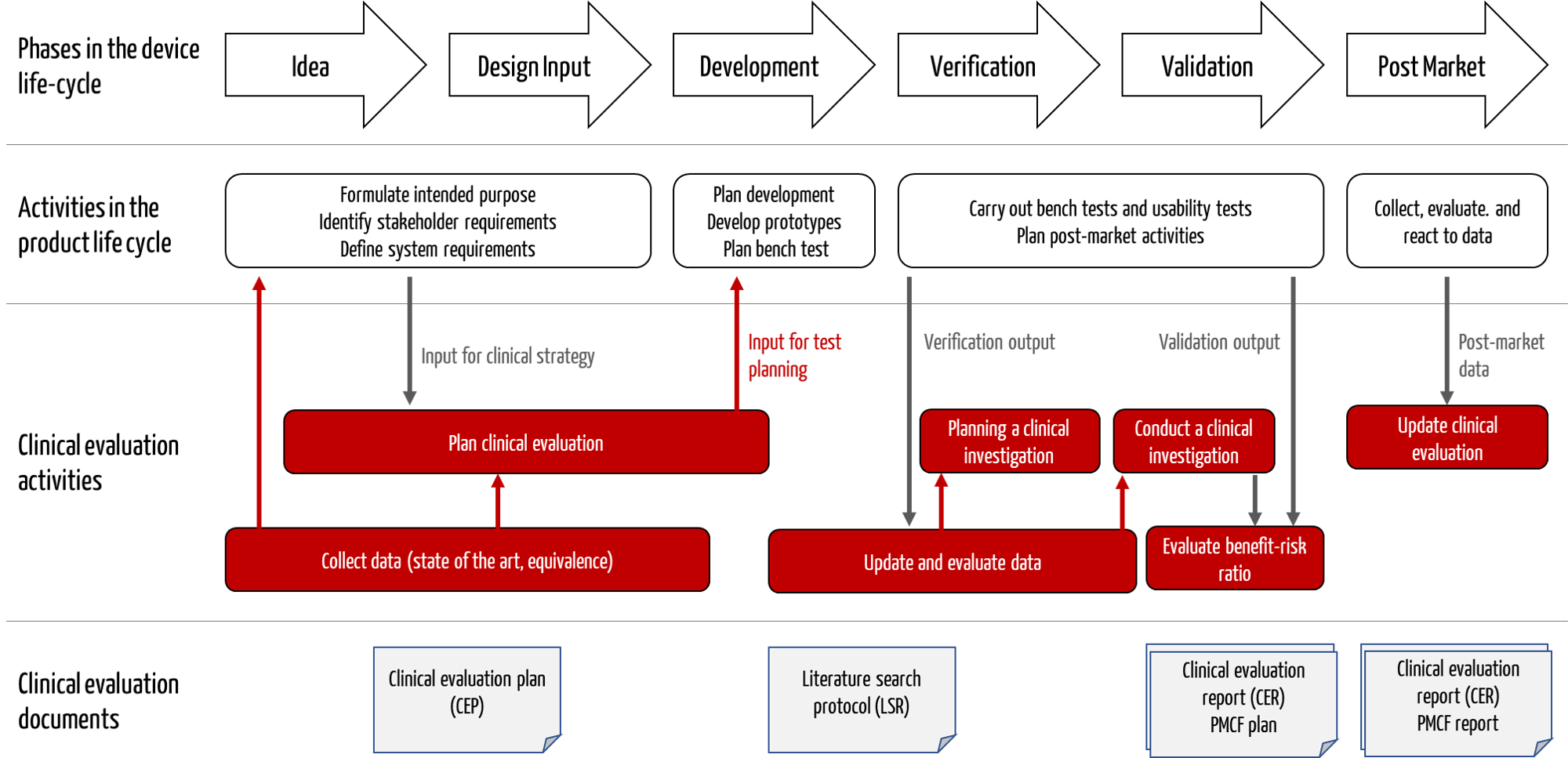
Fig. 1: Clinical evaluation activities interact with development and post-market surveillance.
Tip
Do not only involve the clinical affairs department in the clinical evaluation but also request activities from other competencies such as development, product management, and physicians.
c) Advantages of a professional clinical evaluation
Manufacturers benefit from a professional and legally compliant clinical evaluation:
- Avoid the hassle with reviews, audits, and approvals
The clinical evaluation file is the focus of almost all auditors and reviewers. Deviations are the rule rather than the exception, but they can be avoided.
- Minimize development risks
During clinical evaluation, the manufacturer must research the state of the art at the beginning of development, including the competition and the performance of the devices. This impacts whether clinical investigations are necessary, which often delay the development project by years. Development projects regularly fail because these factors parameters are considered too late.
- Bringing devices to market faster
Rapid development and smooth approval lead to rapid placing in the market and, thus, to early sales.
- Improve patient safety
By definition, clinical evaluation examines the quality of devices, i.e., their safety, performance, and clinical benefit.
- Avoid damage to the image and costs for recalls
High-quality devices are suitable for the company’s image and reduce the costs of product recalls.
- Targeting marketing communication
The clinical evaluation ensures that the intended purpose and intended use are precisely formulated. This forms the basis and framework for coherent and legally compliant marketing communication.
2. Regulatory requirements for clinical evaluation
a) MEDDEV 2.7/1 revision 4
The Medical Device Directive MDD already required a clinical evaluation, albeit somewhat vaguely. This is why MEDDEV Guideline 2.7/1 (revision 4 since 2016) exists, which provides more detailed clinical evaluation instructions.
Although the guideline is not legally binding, it has been the gold standard and reference for the content and structure of clinical evaluations. Auditors still require compliance.
The MDCG guidelines also refer to MEDDEV 2.7/1 Rev. 4, which, therefore, loses none of its relevance.
b) MDR
The MDR dedicates Article 61 and Annex XIV (Part A) to clinical evaluation. Both make many of the requirements of MEDDEV 2.7.1, Rev. 4, legally binding.
c) MDCG guidelines
In addition, the Medical Device Coordination Group (MDCG) has published several guidelines on clinical evaluation. Table 1 provides an overview.
d) Summary
| Regulation/Guideline |
Type / Relevance / How does it help you? |
Prio |
| MDR (2017/745) |
Defines the regulatory requirements that all manufacturers must fulfill. It gives initial instructions on how to carry out the clinical evaluation. |
1 |
| MEDDEV 2.7/1 rev. 4 |
Provides concrete instructions, like a “cookbook,” on how to structure and carry out the clinical evaluation process. |
2-3 |
| MDCG 2020-1 |
Provides a detailed interpretation of the MDR regarding the clinical evaluation of software as a medical device. |
2 |
| MDCG 2020-5 |
Provides a detailed interpretation of the MDR regarding clinical evaluation based on equivalence to other devices. |
2 |
| MDCG 2020-6 |
Gives a detailed interpretation of the MDR regarding the clinical evaluation of existing products. |
2 |
| MDCG 2020-13 |
Is the “checklist” (the CEAR template) that auditors use to assess the clinical evaluation.
(CEAR = Clinical Evaluation Assessment Report) |
2 |
| IMDRF MDCE WG/N56 |
A “quick guide” with specific instructions, shorter than the MEDDEV and not a gold standard, but with helpful tips on literature assessment. |
4 |
Tab. 1: The relevant regulations and guidelines for clinical evaluation
4. Further articles
a) Articles on individual activities
The following articles provide assistance with the activities listed:
b) Articles on clinical investigations
Manufacturers must conduct a clinical investigation if they do not have sufficient clinical data.
c) Articles on regulatory documents and other topics
- MDCG 2020-5: The end of the equivalence route in clinical evaluation?
- MDCG 2020-6: Requirements for clinical data for legacy devices
- Selecting Health Technology Assessment (HTA) for software
5. Support with the clinical evaluation
Do you still have questions about clinical evaluation? Then, benefit from free micro-consulting.
The experts at the Johner Institute are specialized in clinical evaluations and are happy to
- support you with your clinical strategy,
- provide you with clarity about the status of your clinical evaluation using a quick check,
- prepare, write, and review clinical evaluations,
- discuss deviation reports with notified bodies, and
- plan clinical investigations, and perform case number calculations.
Contact us for the quickest way to achieve an accurate and compliant clinical evaluation.
Contact us
FAQ on clinical evaluation
Do class I medical devices require a clinical evaluation?
Clinical evaluation for class I medical devices
All medical devices must meet the basic requirements set out in the MDR. The requirement for a clinical evaluation plays a central (see also MDR, Article 5, Section (3)).
But, in the case of class I devices, the clinical evaluation is not reviewed by a notified body, nor is the entire technical documentation. However, the national authority can ask for it at any time.
Does the MDR allow clinical data to be omitted?
Omission of clinical data
The MDR only allows clinical data to be omitted from the clinical evaluation under particular conditions: The MDR allows this exception in Article 61, sentence (10) if the evidence using clinical data is deemed unsuitable or inappropriate (“is not deemed appropriate”). This waiver of clinical data must be justified in an understandable manner
For example, aids (e.g., oral spatulas) or tools (e.g., scalpels, dental drills) can often be evaluated without product-specific clinical data, as a solitary clinical investigation does not appear appropriate or ethically justifiable. Of course, this must be justified understandably.
For the justification, you can, for example, use general clinical data in the state of the art to demonstrate that your device is based on a proven technology that corresponds to the recognized state of the art and generally undergoes little change. Ideally, product standards are available in which performance parameters are specified. By complying with them, safety can be guaranteed. The benefits can be demonstrated, for example, using guidelines issued by medical societies. The MDCG only describes this possibility in the document MDCG 2020-6 for legacy devices. However, the basic idea is also applicable if you add such a device to your portfolio and approve it for the first time.
Caution!
Note that omitting clinical data does not mean that you do not need to subject your device to user inspection before placing it on the market. In most cases, you will need summative tests, a so-called “usability test,” to validate usability.
Do equivalent devices avoid a clinical investigation?
No clinical investigation for equivalence products
In principle, yes, but the MDR requirements for the equivalence criteria are so high that they can almost only be met in the case of in-house predecessor products. The reason is that you have to demonstrate very detailed knowledge of the other devices.
For class III and implantable devices, the MDR even explicitly requires contracts between manufacturers that provide complete insight into the technical documentation.
But also, if you have predecessor products, you must check very carefully. A device is only considered equivalent if you can prove technical as well as biological and clinical equivalence. A change to the material or coating, therefore, quickly leads to a loss of biological equivalence, an extension of the indications leads to a loss of clinical equivalence, etc.
You do not automatically need a study for every new device. Investigate whether clinical data is necessary or suitable for your device to demonstrate compliance with the basic requirements for performance and safety.
Further information
Read more about equivalence requirements here.
Is the clinical evaluation a document?
Document or process
The clinical evaluation is not a document but a process. The MDCG writes about this in the document MDCG 2020-13:
„ … the clinical evaluation report (CER) and the related clinical evaluation that was conducted – a core requirement of the Medical Device Regulation (EU) 2017/745 (MDR).”
In addition to the clinical evaluation plan, which is already clearly required in the MDR, the MDCG 2020-13 document expands the clinical evaluation file to include a plan and a report on the literature search.
Read more about how these two documents are created here.
When is the clinical evaluation written? At the end of product development?
Time of creation
No! The clinical evaluation process cannot be started early enough. After all, it is not only important for planning but also an essential prerequisite for risk management. Only the clinical evaluation justifies the assumptions made in the risk management file regarding the benefit and, thus, the acceptance of a certain risk-benefit ratio. The clinical evaluation must also confirm the assumptions made in the risk management file regarding the clinical risks.
However, the final clinical evaluation report is one of the last documents to be completed for the approval of a medical device.
How does the clinical evaluation interact with other documents? Does it reference them but is not referenced by them?
Interaction with other documents
The clinical evaluation is not a source document for product information but a summary and assessment. The clinical evaluation documents contain many references to other documents.
The clinical evaluation is not a source document for product information but a summary and assessment. The clinical evaluation documents contain many references to other documents.
Tip
The Clinical Evaluation Report (CER) should be a stand-alone document that can be read independently. Refer to document MDCG 2020-13, the template for the CER that the auditor or reviewer must prepare to see what information the notified body requires to review your clinical evaluation.
