As part of its “Vision 2030,” Saudi Arabia is planning to expand its healthcare infrastructure. Does this offer medical device manufacturers interesting growth opportunities? And with increased approval requirements, is the effort for approval worth it?
Find out in this article how to
- prepare,
- carry out,
- and maintain
approval in Saudi Arabia.
1. An interesting market for medical device manufacturers?
a) A big market
Saudi Arabia has a population of over 34 million. That’s about twice the population of Austria and Switzerland combined. However, healthcare expenditure in Saudi Arabia was only 3.3% of GDP in 2006. In Austria, this figure was approximately three times higher (10.3%).
Saudi Arabia’s high level of dependence on oil also represents a risk for long-term investments. However, the “Vision 2030” project aims to reduce this dependency.
b) Strategic investments in the healthcare market
Part of this initiative explicitly aims to transform the healthcare system. It also aims to promote health tourism. In this context, the Saudi Arabian Medical Attaché in the United States and Canada sais:
The government [of Saudi Arabia] is investing a lot of money in healthcare; the budget for just the Ministry of Health is about $10 billion USD. That does not include the National Guard hospitals, Ministry of Defense hospitals and we have 26 universities, most of those have hospitals.
Source
c) Conclusion
According to the Global Gender Gap Report, Saudi Arabia is one of the last places in the world when it comes to human rights, freedom of expression, and equality. At the same time, Saudi Arabia is a country that already spends a lot of money on healthcare and will continue to increase this spending, making it an interesting market for medical device manufacturers.
However, manufacturers who want to sell their devices in the country first have to meet the regulatory requirements, i.e., they have to have their devices approved in Saudi Arabia.
In this article, you will get information on preparing, carrying out, and maintaining approval in Saudi Arabia. Furthermore, you will learn about the advantages and disadvantages of the SFDA’s alignment with MDR and IVDR.
2. Registration of medical devices in Saudi Arabia
a) Saudi Food and Drug Administration
Medical devices can only be placed on the market in Saudi Arabia if they have been registered with the SFDA (Saudi Food and Drug Administration) and they comply with the Medical Devices Law. The authority is responsible for the registration and monitoring of medical devices.
b) New challenges and benefits for manufacturers from the EU
The SFDA requires that all devices sold in Saudi Arabia be approved via a Medical Devices Marketing Authorization (MDMA). Chapter 4 of this article explains how this works.
Existing approvals from other countries are no longer recognized, which did contribute to simplified registration in Saudi Arabia. The good (but also bad) news is that the SFDA’s current requirements are mostly based on the MDR (and IVDR).
The SFDA supports manufacturers by publishing their requirements for medical devices and IVDs on their website and translating (almost) all of their regulations and laws into English. However, their complexity and the high number of publications regularly cause problems for manufacturers.
c) The most important laws and guidelines
The Saudi Arabian authority published the most important requirements in the “Medical Devices Law.” This regulation contains general requirements for medical devices, their authorization, and market surveillance.
Further regulations specify the requirements of this superior law in more detail. In addition, the SFDA regularly publishes guidelines on specific topics such as quality management systems, UDI, or software.
Tab. 1 provides an overview of the most important requirements.
| Document | Contents |
| Medical Devices Law | General requirements for medical devices, their authorization, and market surveillance. The law is supplemented by regulations (MDS-REQ) and guidelines (MDS-G). |
| MDS-REQ 1 Requirements for Medical Devices Marketing Authorization | Describes the requirements that devices must meet to obtain an MDMA (Medical Device Marketing Authorization). It contains “Essential Principles of Safety and Performance” for medical devices and IVD medical devices, as well as rules for risk classification. All devices must be registered in accordance with these requirements. |
| MDS-G008 Guidance on Medical Devices Classification | Application examples and explanations of the classification and rules of the MDS-REQ 1 |
| MDS-REQ 5 Requirements for Shipments Clearance of Medical Devices at Ports of Entries | Only devices with valid approval will be released by customs. The document explains which other documents must be enclosed to import devices to Saudi Arabia. |
| MDS-REQ 7 Requirements for Unique Device Identification (UDI) for Medical Devices | Explains how medical devices in Saudi Arabia must be provided with a UDI. From September 2023, all devices must carry a UDI. |
| MDS-REQ 11 Requirements for Post-Market Surveillance of Medical Devices | Contains requirements for market surveillance and for reporting incidents and recalls to the SFDA |
| MDS-REQ 10 Requirements for Inspections and Quality Management System for Medical Devices | Contains requirements for the quality management system and the monitoring of manufacturers by the SFDA. All manufacturers require a certificate in accordance with ISO 13485. |
The documents referenced in Tab. 1 can be downloaded from the SFDA website.
The SFDA is not reinventing the wheel regarding requirements for approval, documentation, and classification: the requirements have many parallels with the European MDR and IVDR. For example, the MDS-REQ 1 contains, among other things, the general safety and performance requirements of the MDR and identical requirements for classification.
d) Slightly different definition of the term medical device
The definition of the term medical device differs in some details from the European definition. Therefore, check whether your device falls under this definition. The term is defined as follows in Article 1 of the Medical Devices Law:
“Medical device means any instrument, apparatus, implement, machine, appliance, implant, in vitro reagent or calibrator, software, material or other similar or related article:
Intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific purpose(s) of:
- Diagnosis, prevention, monitoring, treatment or alleviation of disease,
- Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
- Investigation, replacement, modification, or support of the anatomy or of a physiological process,
- Supporting or sustaining life,
- Control of conception
- Disinfection of medical devices,
- Providing information for medical or diagnostic purposes by means of in vitro examination of specimens derived from the human body; and
which does not achieve its primary intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its intended function by such means.”
IVD medical devices are not defined independently but are included in the above definition. However, they are subject to separate classification rules.
In comparison to the MDR, devices for the prediction and prognosis of diseases are not covered by the Saudi Arabian definition. Instead, devices that support the anatomy or a physiological process are classified as medical devices.
This is in contrast to the classification rules of MDS-G008, which give three product examples whose intended purpose is prognosis and prediction.
In case of doubt, the SFDA should be contacted via an Authorized Representative (AR) to clarify the qualification as a medical device.
Devices without an intended medical purpose that are listed in Annex XVI of the European Medical Device Directive must also be treated as medical devices in Saudi Arabia in accordance with the applicable classification rules (MDS-G008, Chapter 1).
3. Classification
Devices are assigned to one of the classes A to D based on their risk. The class determines the authorization procedure, which is presented in the next chapter.
The classification rules can be found in MDS-REQ 1 and are identical to the 22 rules of MDR. You can transfer your classification accordingly (see Tab. 2).
| Classification in Saudi Arabia | Risk level | Classification according to MDR |
| A | Low | I |
| A – Sterile | Low-medium | Is |
| A – Measuring function | Low-medium | Im |
| A – Reusable surgical instruments | Low-medium | Ir |
| B | Low-medium | IIa |
| C | Medium-high | IIb |
| D | High | III |
The SFDA has published the guideline MDS-G008 for the interpretation of the classification rules. The document contains helpful explanations and supplementary examples of how these rules should be applied to devices.
The SFDA also uses European legislation as its base when it comes to the classification of in vitro diagnostic medical devices: The IVDR’s seven classification rules can also be found in Saudi Arabia.
| Classification in Saudi Arabia | Risk level | Classification according to IVDR |
| A | Low individual risk and low public health risk | A |
| B | Moderate individual risk and/or low public health risk | B |
| C | High individual risk and/or moderate public health risk | C |
| D | High individual risk and high public health risk | D |
4. Successful approval of medical devices
Registration with the SFDA is essentially divided into three steps:
- Preparation
- Submission of the documents
- Maintaining the registration
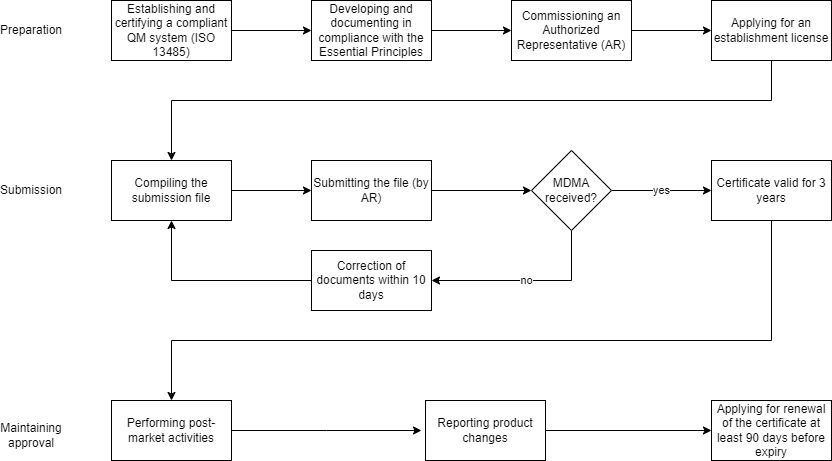
a) The steps to prepare for approval
Step 1: Establishing a QM system
Medical device manufacturers must have a QM system in accordance with ISO 13485. The corresponding certificate and the last audit report from the notified bodies must be submitted to the SFDA. This also applies to manufacturers of class A devices, which are not required to have an ISO 13485 certificate in the EU.
Step 2: Preparing technical documentation
European manufacturers have an advantage when preparing compliant documentation: The technical documentation requirements are contained in Annex 3 (medical devices) and Annex 4 (IVD) of the MDS REQ1 and are based on Annex 2 of the MDR and IVDR, respectively. The SFDA provides a more detailed breakdown of the information it expects from manufacturers in its guidance documents.
The SFDA description is set out in bullet points in the document MDS REQ 1. This is clearer than in the MDR, but it leaves less room for interpretation.
According to the SFDA, the intended purpose must contain at least the following information:
- Main indications of the device
- Disease/disability to be treated
- Patient population to be treated
- Contraindications
The “information on design and manufacturing” is also a good example. The MDR only includes three sub-items. While manufacturers have to think about what information could be required by the MDR to “understand the design stages applied to the device,” the SFDA specifies in eight sub-items what documents and information it specifically expects during design and manufacture.
Step 3: Establishing an Authorized Representative
Manufacturers not based in Saudi Arabia need an Authorized Representative (AR) in the country. This representative must register with the SFDA. The tasks of the AR can be performed by the importer, the distributor, or an independent body.
The tasks of the AR are varied: For example, he is responsible for submitting the submission documents and communicating with the SFDA. He must also ensure that the device complies with the SFDA’s requirements and is labeled accordingly. Only the AR may authorize medical devices.
Your Authorized Representative must also maintain a quality management system.
Step 4: Applying for a Medical Device Establishment License
All manufacturers of medical devices must apply for an Establishment License from the SFDA before devices can be approved. The license is valid for five years and is applied for electronically via the GHAD system. For manufacturers outside Saudi Arabia, the Authorized Representative takes over this activity.
The Establishment License is required not only for manufacturers but also for importers and distributors.
b) The steps for submission
All devices require a Medical Device Marketing Authorization (MDMA) and are authorized via the TFA procedure, regardless of their classification. In the first step, you compile the submission file and submit it to the SFDA via your AR.
Step 1: Compiling the submission file
The SFDA distinguishes between
- a simplified submission for class A devices and
- a regular submission according to the TFA route for all other devices.
Step 1a: Simplified documentation
Devices with a low risk (class A devices) can be submitted with a simplified technical documentation.
In this case, the following documents are required:
- Device description and specifications
- Information on the manufacturer
- Checklist of the essential principles
- Verification documents for compliance with the essential principles
- Risk management file
- PMS plan and PMS report
The complete documentation must always be prepared in accordance with Annex 3 or 4, MDS-REQ 1. The SFDA reserves the right to request additional documentation and grants a deadline of only 10 days for subsequent submission.
Excluded from this simplified procedure are IVD medical devices and class A devices in the following categories:
- As: Sterile
- Am: Measuring function
- Ar: Reusable surgical instruments
- Novel devices
Step 1b: Complete documentation
These and all other devices must be approved via the TFA (Technical File Assessment) procedure.
An existing approval, e.g., a Home Country Approval, is not a prerequisite for the TFA procedure.
As the name suggests, the TFA procedure is based on assessing the technical documentation. The objective of the TFA procedure is to use the technical documentation to check whether the “Essential Principles of Safety and Performance” have been complied with. These can be found in Annex 1 of MDS-REQ 1. You may find them familiar when reading them, as large parts have been taken from Annex 1 of the MDR (including numbering).
For the inspection by the SFDA, you must now submit your complete documentation to MDS-REQ 1 in accordance with Annex 3 (for medical devices) and Annex 4 (for IVD medical devices). This is comparable to the technical file that you submit to your notified bodies for inspection; it contains, for example, the:
- Device description
- Information on the manufacturer
- Information on the design and manufacturing of the device
- Checklist of the “Essential Principles of Safety and Performance”
- Risk management file
- Verification and validation activities performed
- PMS plan
- PMS report (class A) or PSUR (class B, C, and D)
The responsibility for submitting the documents to the SFDA lies with your Authorized Representative. The SFDA will then check your documents within approximately three to four months and issue the written Marketing Authorization after successful inspection.
If the AR cannot answer queries from the SFDA himself, he will contact the manufacturer.
Manufacturers need to stay alert here: if the SFDA does not receive a response to its queries within 60 days, the submission will be deleted. This also applies if the SFDA’s queries are not adequately answered within three cycles. The authority will not refund the fee.
If the submission is successful, you will receive a certificate valid for three years.
c) Maintaining approval
The manufacturer’s obligations do not end with successful approval. Among other things, you must consider the requirements for post-market surveillance and reporting of changes.
- Requirements for the PMS are contained in MDS-REQ 11. It regulates, for example, which information must be reported to the SFDA and which times must be considered.
- Changes to the device must be reported to the SFDA via the AR after 10 or 30 days, depending on the scope.
An application for renewal of the certificate must be submitted at least 90 days before expiry. If the Authorized Representative changes, you have the option of carrying out a license transfer.
5. Conclusion and costs
The SFDA has issued extensive laws and guidelines on medical devices. They are strongly oriented towards European legislation. Whether this has done them a favor remains to be seen.
The approval process, duration, and registration cost depend on the device’s class. Additional costs arise from your representative’s annual fee.
| Class A* | Class Am, Ar, As | Class B | Class C | Class D | |
| SFDA fee [SR] | 15,000 | 15,000 | 19,000 | 21,000 | 23,000 |
| SFDA inspection times | Few weeks | ~ 3–4 months | ~ 3–4 months | ~ 3–4 months | ~ 3–4 months |
| Validity | 3 years | 3 years | 3 years | 3 years | 3 years |
Our team will help you get your medical devices approved in Saudi Arabia. We will be happy to answer your questions; as part of our micro-consulting service, even free of charge.
Change history:
- 2025-02-26: Revision of chapter 4b
- 2023-06-26: Entire revision to incorporate the amended legislation and requirements under the Medical Devices Law
- 2020-10-07: Insertion of the notes in chapters 4.4 and 6 and addition of the column for class Am, Ar, As in chapter 8


