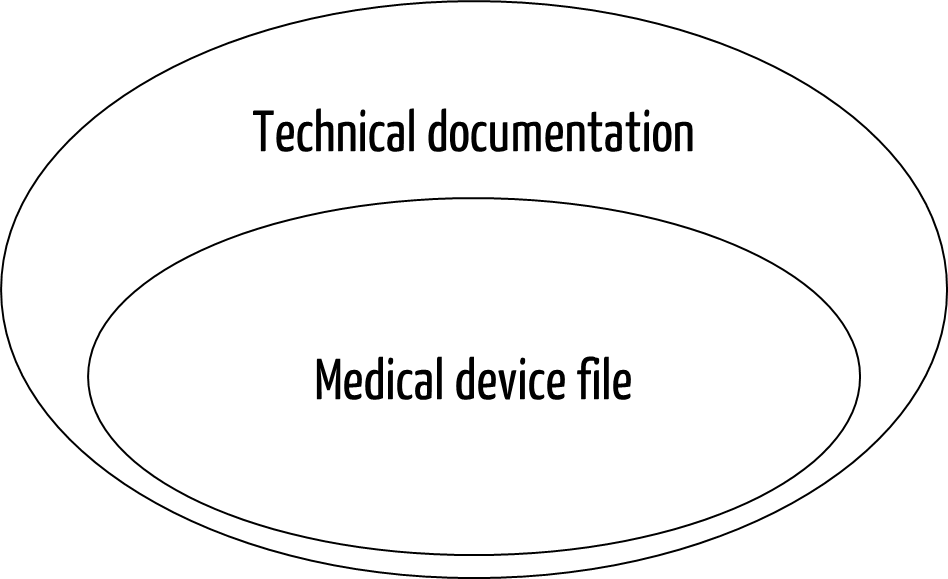
ISO 13485 requires a medical device file for each medical device type or medical device family. Many manufacturers think that the medical device file is the same as the technical documentation required by the MDR or IVDR. But that isn’t entirely true.
Does one of the three files required by the FDA (DMR, DHF, DHR) match the medical device file? It’s time for a comparison to end unnecessary discussions within your team and with auditors.
Free download included.
1. The medical device file: What ISO 13485 aims for and demands
ISO 13485 requires a medical device file since the 2016 edition. The aim is to ensure that manufacturers create (and maintain) all documents required to demonstrate compliance with the regulatory requirements.
The standard talks about “compliance with applicable regulatory requirements.” Herein, it refers to, for example, the MDR and IVDR for medical devices placed on the market in Europe. For devices regulated by the FDA, it corresponds to, for example, the requirements of 21 CFR part 820.
However, ISO 13485 is not limited to referring to other regulations. In fact, it defines which documents the medical device file must (at least) contain, regardless of the jurisdiction.
2. Medical device file versus technical documentation versus “FDA files”
a) MDR, IVDR: Technical documentation
Annex II of the Medical Device Regulation (MDR) and Annex II of the EU Regulation on In Vitro Diagnostic Medical Devices (IVDR) both specify the requirements for the technical documentation. The Post-Market Surveillance (PMS) plans defined in Annex III in both regulations are also considered part of the technical documentation.
Read more on the subject of the technical documentation here.
b) FDA: DHF, DMR, DHR
The FDA distinguishes between three different files:
- Design History File (DHF) according to 21 CFR part 820.30
- Device Master Record (DMR) according to 21 CFR part 820.181
- Device History Record (DHR) according to 21 CFR part 820.184
c) Comparison of the files
The following table compares these requirements.
| document | medical device file according to ISO 13485 | ref. ISO 13485 | technical documentation according to MDR/IVDR | ref. MDR | FDA: DMR, DHF, DHR |
| general description of the device | X | 4.2.3 a) | X (incl. names) | II.1.1.a) | |
| variants | X*) | X | II.1.1. i) | ||
| language versions | X*) | X | |||
| intended accessories and combinations with other devices | X*) | X | II.1.1. h) | ||
| intended purpose and intended use | X | 4.2.3 a) | X (incl. user, patient group, (contra-) indications) | II.1.1 a), c) | 21 CFR part 820.30 |
| warnings | X | II.1.1. c) | |||
| qualification as medical device | X | II.1.1.e) | |||
| classification | X*) | X | II.1.1. f) | ||
| mode of action, scientifically demonstrated if necessary, functionality | X | II.1.1. d) | |||
| description of novel features | X | II.1.1 g) | |||
| reference to previous and similar devices | X | II.1.2. a), b) | e.g., for 510(k) approval | ||
| description of the development stages | X | II.3. a) | 21 CFR part 820.30 b) | ||
| device specifications, e.g., drawings, calculations, components | X | 4.2.3 b) | X (incl. dimensions, performance attributes) | II.1.1. j), l) | 21 CFR part 820.181 a) |
| software specifications | X*) | X | II.6.1 b) | 21 CFR part 820.181 a) | |
| relevant GSPR | X | II.4. a) | |||
| materials used, design drawings | X*) | X (incl. those in contact with the body) | II.1.1 j) | 21 CFR part 820.181 a) | |
| medicinal products used | X*) | X | II.1.1. k), II.6.2. a) | ||
| tissues of human or animal origin used | X | II.6.2 b) | |||
| toxicity, biological safety, and biocompatibility | X*) | X | II.6.1 b), II.6.2. d) | (21 CFR part 820.181 c)), 21 CFR part 820.184 d) | |
| physical, chemical, microbiological, electrical safety | X | II.6.1 b) | (21 CFR part 820.181 c)), 21 CFR part 820.184 d) | ||
| absorption, metabolism, excretion, interaction, tolerance of substances in the body | X | II.6.2 c) | |||
| lifetime, stability, shelf life | X | II.6.1 b) | 21 CFR part 820.184 d) | ||
| measurement accuracy (in case of measuring function) | X | II.6.2.ef) | 21 CFR part 820.184 d) | ||
| interoperability, compatibility | X | II.6.2.g) | (21 CFR part 820.181 c)) | ||
| software V&V results | X | II.6.1 b) | 21 CFR part 820.30 | ||
| specifications for manufacture, e.g., requirements for facilities, infrastructure, production methods | X | 4.2.3 c) | (incl. adjuvants, processes, validation) | II.3. b) | 21 CFR part 820,181 b) |
| quality assurance requirements, e.g., procedures, equipment, acceptance criteria | X*) | X | II.6.1 (and others) | 21 CFR part 820.181 c) | |
| details of suppliers and contractors with their activities | X | II.3. c) | |||
| packaging specifications, e.g., methods, processes, materials | X | 4.2.3 c) | X | 21 CFR part 820.181 d) | |
| specifications (procedures) for measuring and monitoring | X | 4.2.3 d) | (21 CFR part 820.181 c)) | ||
| requirements for Post-Market Surveillance (PMS), particular the PMS Plan | X | III | |||
| design outputs | X*) | X | II.6. (and others) | ||
| tests, e.g., laboratory, simulation, animal testing | X | II.6.1 a) | (21 CFR part 820.181a)), 21 CFR part 820.184 d) | ||
| clinical data | X*) | X | II.6.1 c) | ||
| clinical evaluation, incl. CER and CEP | X | II.6.1 c) | |||
| PMCF plan | X | II.6.1 d) | |||
| risk management, e.g., risk analysis, risk-minimizing measures, list of residual risks, risk-benefit evaluation | X*) | X | II.5. a), b) | ||
| storage requirements | X | 4.2.3 c) | |||
| specifications for transport | X*) | 4.2.3 c) | |||
| specifications for distribution | X | 4.2.3 c) | |||
| requirements for installation | X | 4.2.3 e) | 21 CFR part 820.181e) | ||
| requirements for maintenance and servicing | X | 4.2.3 f) | 21 CFR part 820.181d) | ||
| instructions for use | X | 4.2.3 c) | X | II.2. | |
| other labeling | X | 4.2.3 c) | X (packaging) | II.2. | |
| UDI | X*) | X (Basic UDI) | II 1.1. b) | ||
| changes to the device during its lifetime and related V&V | X*) | X | Article 10 (and others) | ||
| list of standards (or “other methods”) used to demonstrate conformity | X*) | X | II.4. b), c) | ||
| (documents to) demonstrate conformity with GSPR | X*) | X | II.4. d) |
Careful! Just because, a cell is not marked with an X, do not conclude that the regulation does not require the document. It just doesn’t mention it explicitly.
The MDR and IVDR insist on more documents for the technical documentation than ISO 13485 does for the medical device file. For example, test results and PMS plans are mandatory components of the technical documentation but not of the medical device file. Put simply, the technical documentation is a superset of the medical device file.
If you compare the medical device file to the “FDA files”, it is most similar to the device master record.
You can download this table as an Excel file: Table-Medical Device File-incl-IVDR-V3
Do you want to ensure that your technical documentation meets the requirements of the MDR/IVDR?
Our e-learning course demonstrates step-by-step how to create a compliant structure for your documentation. You will learn how to check your documents for completeness, consistency, and conformity. After completing the course, you can review your documentation and ensure it is compliant.
3. One medical device file for multiple devices?
a) Medical device family vs. generic device group
ISO 13485 allows to create a medical device file for several medical devices of the same medical device family.
“Group of medical devices manufactured by or for the same organization and having the same basic design and performance characteristics related to safety, intended use, and function.”
ISO 13485 Chapter 3.12
A look at the definition of “generic device group” in the MDR reveals that the two “groups” are not identical:
“Set of devices having the same or similar intended purposes or a commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics.”
MDR Article 2(7)
Conclusion: According to the MDR, the generic device group is somewhat broader defined than the medical device family. The medical device family must not include devices whose intended purpose is only similar.
b) Common file for the same medical device family and the same Basic UDI-DI
The MDR and the IVDR allow to create a common technical documentation for several medical devices with the same Basic UDI-DI. Note, with the same Basic UDI-DI, not for a generic device group! A common Basic UDI-DI, in turn, requires that
- the devices have the same intended purpose,
- their layout, i.e., the design, is essentially the same, and
- the manufacturing, i.e., production, is essentially the same.
If you compare the requirements for grouping, you will see that MDR and IVDR (see also MDCG 2018-3) further restrict the boundaries compared to ISO 13485:
| consistency regarding | ISO 13485 | MDR, IVDR (Basic-UDI-DI) |
| intended purpose | X | X |
| design, performance characteristics | X (only regarding safety) | X |
| manufacturing, production | X |
Thus, the Basic UDI-DI, rather than the medical device file, should determine for which devices manufacturers may create a common medical device file.
4. The medical device file as a file?
ISO 13485, like the EU regulations, does not insist on a physical file. Neither do manufacturers have to fill redundant file folders with paper, nor do they have to store digital documents redundantly in directories.
Instead, manufacturers are well advised to have a (digital) “introductory document” that includes the following:
- context, especially a brief description and identification of the device,
- table of contents (here, you can follow common formats such as STED),
- a short summary of the respective documents, and
- link to these documents to identify the details.
The objective should be to provide readers (colleagues, auditors) an overview and efficient navigation to the desired documents.
5. Conclusion and tips
a) Criticism
Since the 2016 edition, ISO 13485 has insisted on a medical device file. It is debatable how useful this new concept is:
- Unnecessary
Manufacturers must generate technical documentation or FDA-compliant files (DMR, DHF, DHR) anyway. These are the superset of the medical device file. ISO 13485 additionally refers to these regulations in the same chapter. - Confusing
ISO 13485 introduces the term “medical device family.” This term creates confusion with the term “generic medical device group.” Even regulatory documents do not use both terms precisely. - Unspecific
The content required by the standard is too generic to be helpful to manufacturers. As a result, auditors use the more specific practical guide, which seems more aligned with the MDR/IVDR. It even refers to “General Safety and Performance Requirements.” - Expensive
To remain on par with auditors, manufacturers do not only have to buy the standard but also the practical guide. Admittedly, the costs are manageable.
b) Benefits
Manufacturers are allowed to create a common medical device file for different devices in a medical device family. This can save work and is good news.
However, the MDR and IVDR permit a common technical documentation, only for devices with the same Basic UDI-DI. Devices with the same Basic UDI-DI must be “more similar” to each other than devices in the same medical device family.
There is also hope that the medical device file concept paved the way for the FDA to replace its 21 CFR part 820 by ISO 13485. That would be a real simplification. The medical device file is already the equivalent of the device master record.
c) Next steps
The Johner Institute recommends that manufacturers:
- Purchase the practical guide and use it during internal audits (not only regarding the medical device file).
- Download the Excel file and use it as a checklist to verify the completeness of the files.
- Check if files of very similar devices can be merged to simplify their future maintenance.
Do you have questions regarding the medical device file? Would you like someone to check your files for completeness or help you revise them so that the following audit and approval will go smoothly?
Get in touch via our web form or benefit from the free micro-consulting.


