The use of CMR substances is strictly regulated. The MDR also regulates CMR substances and places strict requirements on medical device manufacturers.
This article helps to fulfill these requirements.
1. CMR substances: The challenges
CMR stands for:
- Carcinogenic (cancer-causing)
- Mutagenic (genetically modifying)
- Reprotoxic (toxic to reproduction)
CMR substances are challenging for medical device manufacturers:
- CMR substances pose significant risks to patient safety.
- However, the adverse effects on patients often manifest only in the long term and usually cannot be covered by post-market surveillance.
- Identifying which substances qualify as CMR (carcinogenic, mutagenic, or toxic to reproduction) is not immediately obvious.
- The legal requirements are particularly stringent.
- Demonstrating the absence of CMR substances, and thereby ensuring compliance, proves challenging.
The following chapters will help you to overcome these challenges.
2. Classification as a CMR substance
Manufacturers must be aware of the substances contained in and emitted by their devices, as well as whether these substances are classified as carcinogenic, mutagenic, or toxic to reproduction.
Databases help with this classification.
a. Toxicological databases
Toxicology databases contain data from in vitro and in vivo experiments that provide information on whether a substance has a carcinogenic, mutagenic, or reprotoxic properties.
Examples of toxicology databases are:
The QSAR Toolbox can query various databases simultaneously.
b. Annex of the CLP Regulation
The CLP Regulation contains a list of CMR substances with their official classification in Table 3 of Annex VI.
The regulation’s official title is “Regulation (EC) No 1272/2008 of the European Parliament and of the Council of 16 December 2008 on classification, labelling and packaging of substances and mixtures.”
Databases are available to help with this classification. Please note that the database classifications are often based on different study data, so different conclusions/classifications may arise.
In cases of contradictory data, consulting an expert with toxicological expertise is advisable to help clarify the assessment.
c. European Commission list
The EU has published an overview of CMR substances, although this focuses on cosmetics. However, these substances can also be found in medical devices and are therefore relevant.
d. IFA CMR list (German)
The Institute for Occupational Safety and Health of the German Social Accident Insurance (IFA) has published a CMR list. This comprehensive list contains substances classified as CMR and is updated regularly.
e. IARC Monographs
The EU website contains a list from the IARC (International Agency for Research on Cancer).
3. Origin of CMR substances in medical devices
There are many different ways in which CMR substances can be included in devices. It is not always possible to clearly trace their origins. Potential sources include:
- CMR substances are already present in the starting material.
- The substances are residues of process chemicals.
- The CMR substances are produced during production, e.g., when heated or through reactions with other materials.
4. Detection of CMR substances
The detection and evaluation of CMR substances take place in several steps.
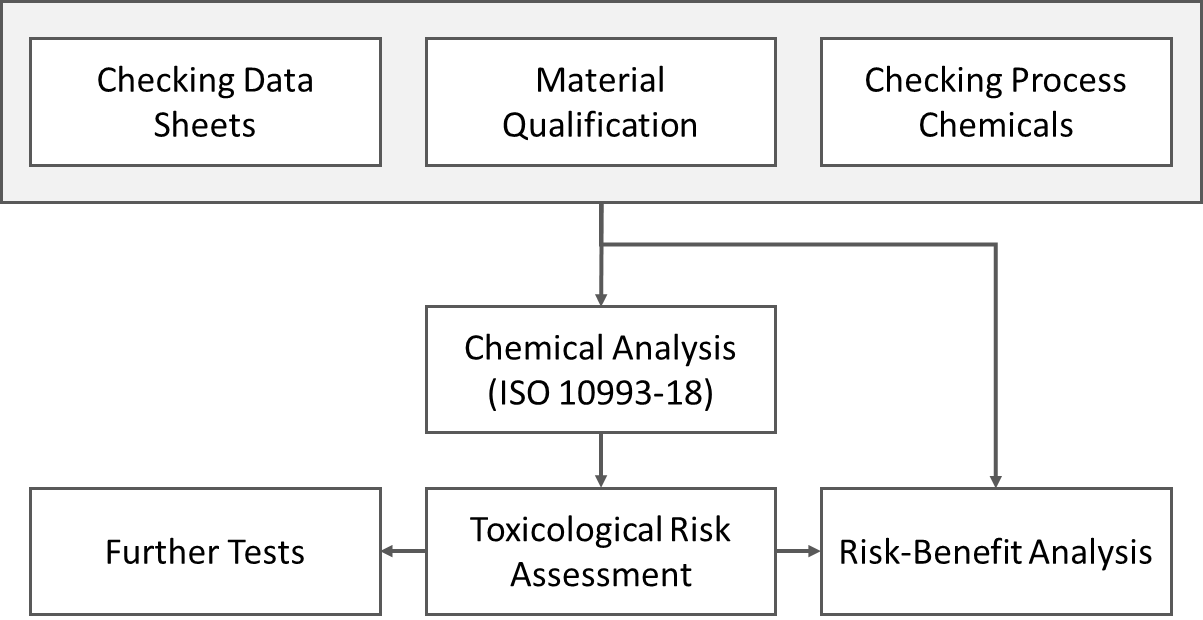
Step 1: Check the data sheets
In the first step, manufacturers should check the starting materials’ data sheets and ensure the material’s suitability by means of a material qualification. The same applies to the data sheets of the process chemicals.
Step 2: Perform chemical analysis
The chemical analysis ultimately clarifies whether the final device releases CMR substances. This analysis should always be included in the tests required for biocompatibility in accordance with ISO 10993-18 for the chemical characterization of the device. It is essential that an expert with toxicological expertise evaluates the results.
Step 3: Toxicological risk assessment and further tests
A toxicological assessment, based on the results of chemical characterization, can clarify the risk associated with any potential release of CMR substances by comparing these results with relevant limit values. This assessment determines whether additional testing is necessary, such as tests for genotoxicity, carcinogenicity, and reproductive toxicity, in accordance with ISO 10993-3.
5. Regulatory requirements
a. MDR requirements
MDR, Chapter II, 10.4.1
Devices that are invasive or directly interact with the body for administering or collecting medicines or body fluids, or for storing body fluids, may contain CMR substances in concentrations above 0.1% weight by weight (w/w) only if it is justified.
MDR, Chapter II, 10.4.2
The justification for CMR substances must include:
- An analysis of potential exposure
- An analysis of available alternatives, including scientific assessments and studies
- A justification as to why alternatives may be inappropriate, taking into account the device’s functionality and its benefit-risk profile
- Consideration of special patient groups (children, pregnant women, breastfeeding women).
If available, current guidelines of scientific committees should be considered.
b. ISO 10993 family
As part of the biological safety assessment according to ISO 10993-1, the endpoints of genotoxicity (including the mutagenic potential of substances), carcinogenicity, and reproductive and developmental toxicity must be addressed.
In the context of toxicological risk assessment, 10993-17 provides information on the risk evaluation of carcinogenic substances. These evaluations should always be carried out by experts with the appropriate toxicological expertise.
c. Further requirements for CMR substances
Other regulations have an indirect effect: The REACH regulation regulates the handling of substances of very high concern (SVHC), which includes many CMR substances. The CLP regulation regulates chemical substances’ classification, labeling, and packaging. In addition, national regulations must be observed (e.g., legislation on occupational safety and the Hazardous Substances Ordinance).
If you cannot avoid using CMR substances in your device, ensure the documentation (label, IFU) is adapted accordingly.
The legal requirements are described in the article on labeling for medical devices.
6. Tips for meeting the requirements for CMR substances
Tip 1: Choose CMR-free substances
Use only starting materials for which you receive comprehensive data sheets. Check and evaluate the substances listed, and avoid starting materials that are or contain CMR substances.
Based on this, carry out a material qualification and document why the material is suitable. At this point, consult with technical experts.
Select only materials that are free of CMR substances for all production steps.
If the substances cannot be avoided, keep their concentration below the 0.1% limit. If you cannot meet this limit, have an expert with toxicological expertise conduct an exposure assessment and a risk-benefit analysis.
Tip 2: Perform tests early in the project
Medical device manufacturers frequently relegate the issue of CMR substances to a lower priority, deferring the necessary testing until the late stages of product development. This limits the options for action and often results in high rework costs and project delays.
Early knowledge of the substances often allows for the redesign of the device to avoid or reduce the release of CMR substances from the final device.
Tip 3: Adhere to the ISO 10993 series
Check the biocompatibility of the final device as specified by ISO 10993. This must include a detailed chemical characterization.
Evaluate the results and, if necessary, conduct a toxicological risk assessment.
Tip 4: Ensure appropriate expertise
Make sure that the persons responsible for this have documented expertise. They must also be familiar with toxicological details and able to assess the exposure and its effects on critical patient groups, for example.
7. Conclusion and summary
Understandably, legislators place particularly strict requirements on carcinogenic, mutagenic, and reprotoxic substances.
Therefore, manufacturers should avoid these substances in their medical devices as far as possible and provide evidence that the CMR substances remain below the toxicologically applicable limit values. Nevertheless, a toxicological risk assessment is necessary and places high demands on the competence of those responsible.
The Johner Institute helps medical device manufacturers demonstrate that they meet all legal requirements (including those for CMR substances), enabling the rapid approval of safe medical devices.
- The Johner Institute’s experts pragmatically assess biological safety, carry out chemical characterizations, and evaluate the results.
- They also help evaluate and classify existing test results with regard to the CMR substances detected.
- Our experts provide risk-benefit analyses for devices that contain CMR substances.
- The Johner Institute offers flexible advice on all questions for all devices and all markets.
Contact us, for example, via our contact page, and you will receive immediate support.


