This article describes when and why manufacturers must affix a CE mark, what is meant by CE marking, and what manufacturers must do up to that point.
Colloquially, the terms CE mark, CE label, and CE marking are used interchangeably. The MDR uses the terms CE marking of conformity and CE marking.
1. Meaning of the CE mark
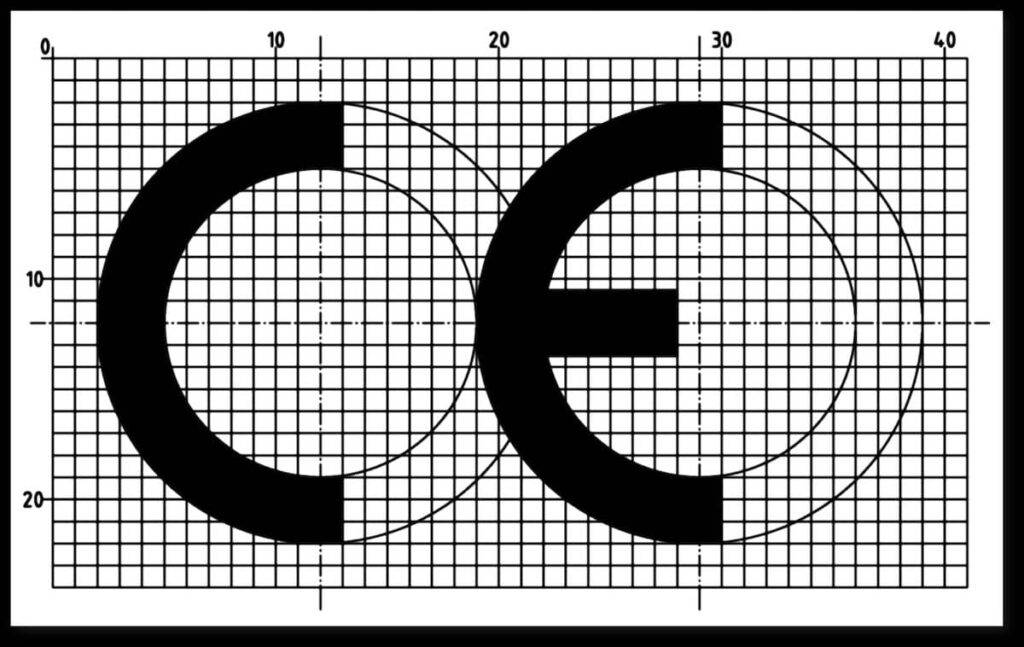
Figure 1 (left): Annex V of MDR and IVDR defines the appearance of the CE marking. With the CE mark, manufacturers express that their device complies with the requirements of a European directive or an EU regulation.
Medical device manufacturers use the CE marking to express that their devices conform to the EU MDR or IVDR regulations. The term CE audit does not exist.
2. Requirements for CE marking
MDR and IVDR require the following from manufacturers regarding CE marking:
- Medical devices must be affixed with the CE marking permanently. (There are exceptions for custom-made devices and investigational devices).
- The CE mark must look as shown in Fig. 1 and be at least 5 mm high.
- If a notified body was involved in the conformity assessment procedure, its number must appear next to the CE mark (see Fig. 2).
- The medical devices must meet the requirements of the regulations (and other regulations and directives, if applicable) before the manufacturer may declare conformity and affix the CE marking.

3. Affixing the CE mark on software
Standalone software (websites, apps, or PC software) can also be a medical device. Therefore, these devices also require CE marking.
Read more about when software counts as a medical device. In most cases, this software then falls into Class IIa or higher. Then, the manufacturer must involve a notified body and indicate its number on the CE mark.
There is not a lot of Class I software anymore.
Manufacturers should affix the CE mark to standalone software, as far as is reasonable and possible, to or at:
- Start or splash screen.
- Help, About
- Website itself
- Accompanying materials, especially instructions for use, manual
There is no regulatory obligation to affix the CE mark in a web or app store.
The requirements of MDR and IVDR apply regardless of whether the offer of, for example, a website is free of charge or not or whether the site is only accessible via a password. Thus, the obligation to affix the CE marking also applies regardless.
4. The way to the CE mark
a) Overview of the phases
The whole process to obtain the CE mark includes several phases (see Fig. 3).
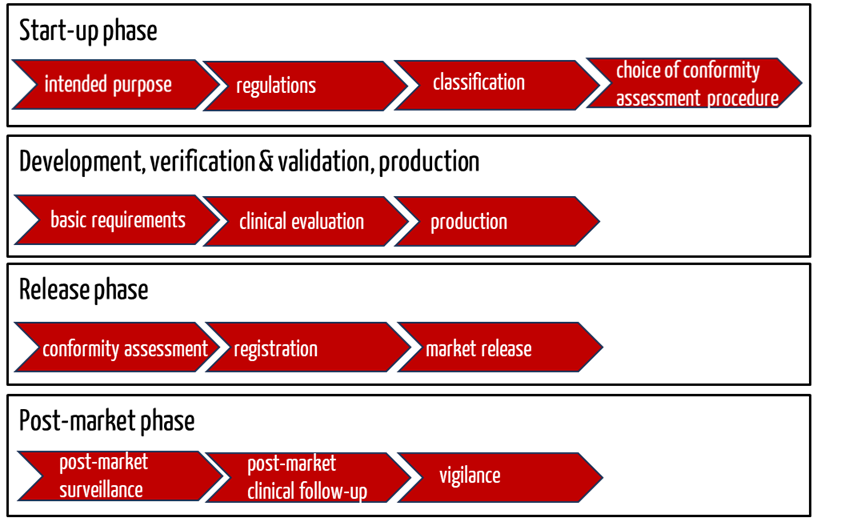
b) Start-up phase: Preparations for the CE mark
- Intended purpose
Formulate the intended purpose of your device. Specify exactly whether and how it is used to diagnose, treat, or monitor disease and injury, who the intended users are, and the intended context of use. - Regulatory requirements
Determine which EU regulations affect your medical device (Medical Device Regulation MDR, IVD Regulation IVDR). - Classification
If the MDR covers the device, use the classification rules in Annex VIII to determine which class your medical device belongs to (I, IIa, IIb, or III).We can assist you with this classification (contact us). - Choice of conformity assessment procedure
Depending on the chosen class, you choose the conformity assessment procedure and may then have to establish a quality management system, which you must have certified according to ISO 13485.With our help, companies always succeed in doing this on the first try.
Thus, there is no “CE certification” and no “CE audit”, but there may be a QM audit and an ISO 13485 certificate or annex certificate (according to the annexes of the MDD or MDR).
c) Development, test and production phase
- General requirements
Develop the medical device according to the requirements of your QM system, if one exists. In any case, it must comply with all basic requirements, e.g., the requirements for risk management, software life cycle processes, usability, and electrical safety.
You prove this by complying with harmonized standards and documenting this with technical documentation.
This Technical Documentation consequently contains:- Risk management file (compliant with ISO 14971)
- Software file (compliant with IEC 62304)
- Suitability for Usability-File (compliant with IEC 62366)
- Electrical safety file (compliant with IEC 60601-1)
- Clinical evaluation
As part of the clinical evaluation, you demonstrate with clinical data that your medical device has no undesirable side effects and meets its intended benefit. Without reliable literature data, you must conduct a clinical investigation. - Production
Finally, you produce, i.e., reproduce your medical devic.
d) Release phase including CE marking
- Declaration of conformity
Declare the conformity of your device with the general requirements. To do this, draw up a declaration of conformity and affix the CE mark to your device. - Registration
Register yourself and your medical device with EUDAMED or BfARM. - Market release
Finally, issue the market release and place your device on the market – with the CE mark.
e) Post-market phase
The final phase continues as long as at least one of your devices is on the market. In this phase, you continuously review feedback from the “field,” respond to incidents, and update your risk management files.
Read more about post-market surveillance and vigilance here.
5. Conclusion and summary
Affixing the CE mark is one of the more manageable tasks that manufacturers must master to bring their medical devices onto the market in compliance with the law and to be allowed to keep them there. The tricky part is to create the conditions for this, i.e., to prove conformity. Nevertheless, this CE marking must also meet the statutory requirements.
Johner Institute supports the manufacturers in the complete process, including preparing the technical documentation and the clinical evaluation.
Change history
- 2023-08-22: Editorial changes only (including spelling errors removed).
- 2023-05-04: Article fundamentally revised, requirements of the MDR described in more detail.
- 2019-06-17: First version of the article


