The NMPA Usability Guidance affects many manufacturers of medical devices and IVDs, as well as manufacturers of combination products, who want to market their products in China.
This article clarifies which devices must comply with the NMPA requirements regarding usability and what these requirements are.
1. Whom the NMPA Usability Guidance concerns
a) Included and excluded device classes
The guidance document of the Chinese regulatory authority NMPA has been applicable since October 8, 2024. It mainly concerns class II and III devices, including IVD and drug-device combination products. However, IVD reagents remain outside the scope of application.
b) Grandfathering
When approvals/registrations are renewed, the NMPA does not insist on additional data from usability engineering.
On the other hand, “grandfathering” does not apply to substantial changes to the users, the use scenarios, or the user interface. In this case, the manufacturers must at least evaluate the changed “aspects” by usability engineering.
The article on the NMPA describes the requirements of the Chinese authority that go beyond usability engineering.
2. Decisions to determine the NMPA requirements
The NMPA’s usability guideline requires manufacturers to check which requirements of its Usability Guidance must be met. These requirements depend on the answers to the following three questions (see Fig. 1):
- Is the device a “high use risk medical device”?
- Is the device novel (or is there a predicate device)?
- Is there sufficient data available from a predicate device?
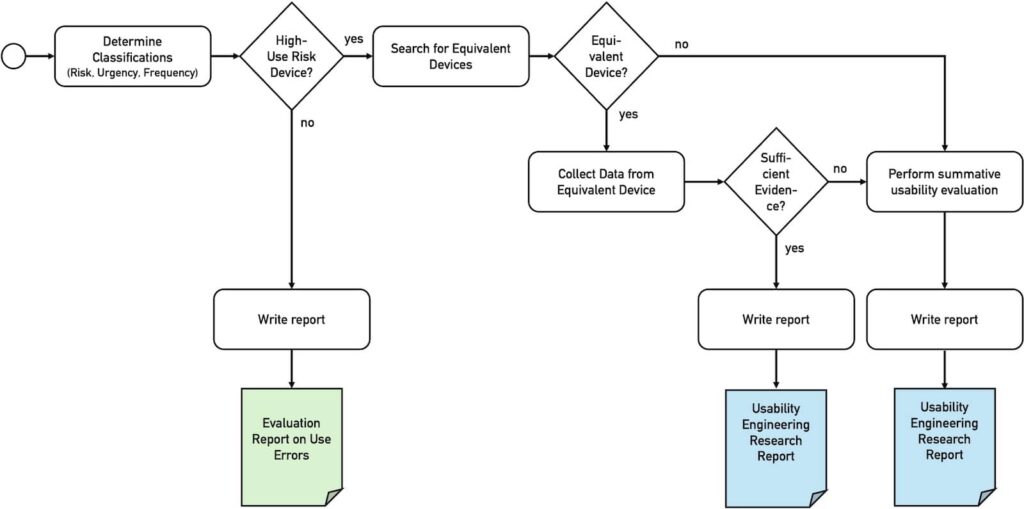
The following sections discuss these three decisions.
2.1 Decision on whether the device is a high use risk medical device
Manufacturers must first determine whether their device is a “high use risk device.” In this context, the NMPA defines three risk classes (see Tab. 1).
| Class | Severity of Possible Harm | Recall Level |
| High Use Risk | Incorrect use may lead to serious injury or death | First |
| Moderate Use Risk | Incorrect use may lead to minor harm | Second |
| Low Use Risk | Incorrect use is unlikely to lead to harm | Third or none |
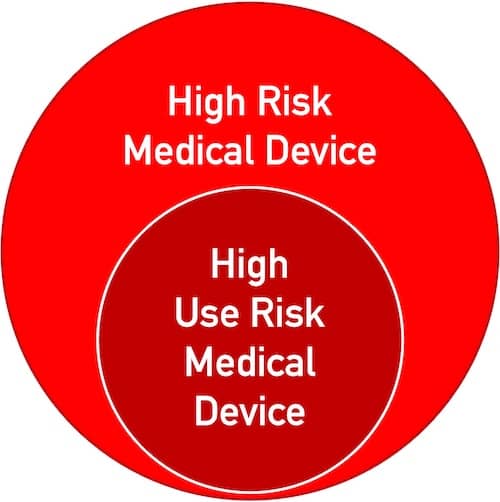
The classification must be based on the “use risks,” not on the general risks that the device can cause. However, only “high risk medical devices” can also be “high use risk medical devices” (see Fig. 2).
For devices already on the market, or in the case of comparable devices already on the market, classification can also be based on “recall levels” instead of the severity of possible harm (see Tab. 1).
Overall, the NMPA requires a categorization of the tasks to be performed with the device according to these three criteria:
- use risk
- urgency
- frequency
Only the first classification (risk) determines the requirements to be met. However, the NMPA expects all classifications to be mentioned in all reports.
In most cases, high use risk medical devices are critical IVD or medical devices that
- are used in a completely new way,
- require a steep learning curve,
- are used by non-professional users, or
- are highly complex to use.
2.2 Decision on whether the device is novel
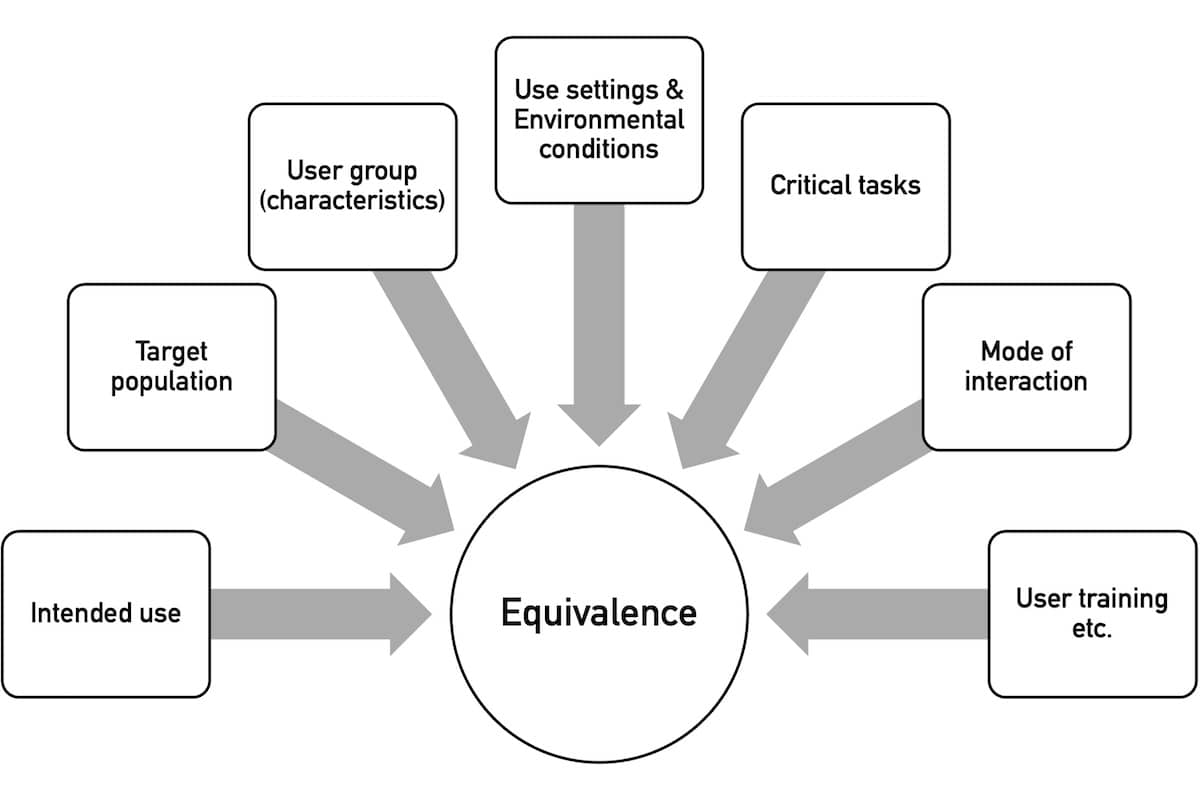
The second step is to decide whether a device is novel or whether there is a comparable predicate device. The NMPA assesses the equivalence using several characteristics (see Fig. 3).
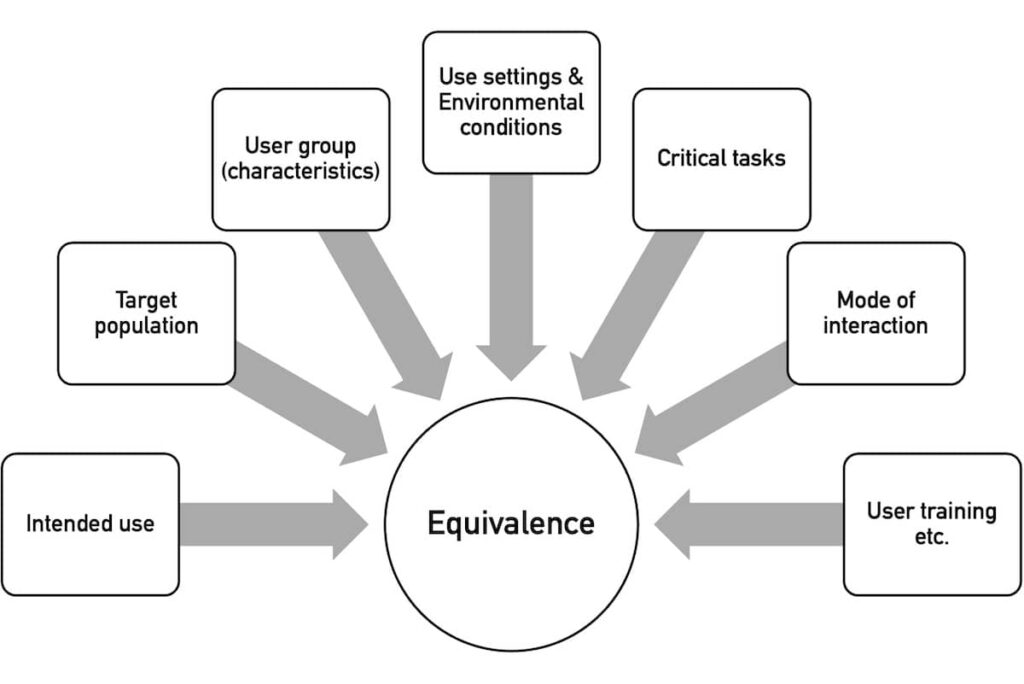
By “use setting,” the NMPA means the use environment, e.g., “outpatient clinic, emergency room, operating room, ward, ambulance, home, and public place.”
“Environmental conditions” refer to the physical environment, such as “space, lighting, temperature, humidity, air pressure, cleanliness, noise, vibration, and radiation.”
For all attributes that are not equivalent, the manufacturers must conduct a usability test (see section 2.3).
When comparing equivalency, manufacturers may only refer to devices registered and marketed in China.
2.3 Decision on whether sufficient data are available
The NMPA insists that the manufacturer proves equivalence regarding usability, i.e., provides sufficient evidence. This includes the following data, for example:
- Results of the usability validation of the predicate device
- If these are not available, an analysis of the post-market usability problems of the predicate device
- Analysis of the post-market usability problems of similar devices
Depending on the comparison result (see “Sufficient Evidence” diamond in Fig. 1), carrying out a usability validation/a usability test in China may be necessary. Tab. 2 describes which activities are required.
| Comparison with the predicate device | No new “use risks” | New “use risks“ |
| No differences to the predicate device | No usability validation necessary | Usability validation necessary for new risks |
| Differences to the predicate device | Usability validation is necessary for the differing aspects | Usability validation necessary for new risks and for the differing aspects |
Suppose the predicate device based on the attributes in Fig. 3 differs (e.g., an additional user group) but poses no new use risks. In that case, the manufacturer must conduct a usability test with this user group in China.
The first line of Tab. 2 distinguishes whether there are new “use risks.”
An analysis of post-market usability problems reveals that a use scenario that was previously not associated with risks is suddenly associated with risks due to new findings from the market. This scenario must then be subjected to a usability test.
3. Requirements of the NMPA Usability Guidance
3.1 Requirements for usability testing
In its Usability Guidance, the NMPA specifies many requirements similar to those of the FDA and IEC 62366-1, but they are not identical. For example, it requires:
- Tests must be carried out in China
- Representative test environment
- At least 15 representative users from each “user group”
- Testing of all critical tasks
- Experienced usability testers, among whom no “Stakeholder” such as developers may be
Neither the EU nor the FDA explicitly prohibits using stakeholders as testers. If a manufacturer does not have a large enough team for NMPA tests, they can use external service providers such as the Johner Institute.
The NMPA generally allows data from other countries to be used. However, approval in the home country is mandatory for approval in China, and the NMPA expects
- a report on “post-market use problems” for similar devices and
- a report highlighting the differences between the requirements in China and the other country and specifying which data is still needed.
The NMPA guideline explicitly states:
“detailed data need to be provided to confirm that the differences between China and foreign countries have no significant impact on the user interface validation” (usability test results from foreign countries).
Since the cultural differences between China and the EU/USA are comparatively large, it is particularly difficult to argue why users should make the same mistakes as in Western countries.
This increases the probability that usability tests will have to be carried out in China.
Usability tests are one of the most important summative evaluation methods that correspond to what the NMPA expects.
3.2 Requirements for the analyzes
Report on Post-Market Use Problems
The report on known post-market problems must include
- what information was searched for (e.g., on which devices),
- when (date, period), how (e.g., search terms), and where (e.g., databases) the search was conducted,
- what the results of the search are (sources, evaluations, conclusions).
Report for Comparison
This report is comparable to other assessments of equivalence, as known from the FDA’s predicate devices.
Read here what predicate devices and substantial equivalence are.
The “Report for Comparison” should also state:
- The objective of the evaluation
- The compared devices
- The selection of the predicate device
- Data suggesting equivalence
- The description of the evaluation path (see Fig. 1)
- Conclusions
- Information about the person who performed the evaluation
Usability Test Report
The contents of the Usability Test Report correspond to what the FDA and IEC 62366-1 expect:
- Test object (device)
- Test subjects
- Test conditions, test environment
- Pass/fail criteria
- Use scenarios and tasks
- Results
- Assessments, conclusions
3.3 Requirements for the documents to be submitted
Depending on the use-risk classification (see Fig. 1), the NMPA expects different documents.
Evaluation Report on Use Errors
The NMPA expects this report for IVD and medical devices with low or moderate “use risks.” It specifies the following structure for this:
- Basic information
- Level of use risk including justification
- Core elements (e.g., users, use scenarios, and user interfaces)
- Analysis of post-market use problems of similar medical devices
- Use risk management
- Conclusion
Usability Engineering Research Report
Manufacturers are required to create a more detailed report for high use risk devices. The NMPA specifies the following structure:
- Basic information
- Level of use risk including justification
- Core elements (e.g., users, use scenarios, and user interfaces)
- Usability engineering process
- User interface requirements specification
- Use risk management
- Verification and validation of the user interface (include materials described in section 3.2 here, depending on evaluation route)
- Traceability analysis of user interface
- User training protocol
- Conclusion
4. Summary and conclusion
The requirements of the NMPA Usability Guidance are mainly consistent with the requirements of the FDA and IEC 62366-1.
Nevertheless, there are some regulatory differences, and in particular, there is an obligation to conduct usability tests in China for devices with high use risk.
Whether the NMPA requirements are intended only to ensure patient safety or foreclose the market cannot be evaluated from the outside.
Once again, manufacturers are faced with the task of creating many market-specific documents (in particular reports), which are largely based on the same usability data. IT systems that generate these approval documents at the push of a button are helpful here.
The Johner Institute offers comprehensive usability services.
- Provision of templates for the reports mentioned above
- Performance of usability tests in its own usability labs in Germany and the USA
- Organization of the performance of usability tests in countries such as China
The Johner Institute also guides IVD and medical device manufacturers through their digital transformation and offers enterprise software for regulatory processes such as approval and market surveillance.
Contact us to discuss how to bring your devices to global markets quickly and with minimal effort. This meeting is non-binding and free of charge.


{kind=link}
{kind=link}