A ‘Request for Information’ can (partially) avoid expensive legal fees. Just ask the authorities directly! This way, you will get a qualified answer, even if it is not free of charge.
Read here to find out
- what questions the FDA answers,
- how to submit a ‘Request for Information’, and
- what the US authority expects from you.
1. What is a ‘Request for Information’?
US law requires its authority – the FDA – to answer questions from manufacturers of medical devices regarding classification within 60 days.
This requirement can be found in the Food, Drug & Cosmetic Act [Article 513(g) (§ 360 c.(g) in the U.S. Code)]:
Within sixty days of the receipt of a written request of any person for information respecting the class in which a device has been classified or the requirements applicable to a device under this Act, the Secretary shall provide such person a written statement of the classification (if any) of such device and the requirements of this Act applicable to the device.
2. What can be clarified with a ‘Request for Information’
2.1 Classification
You can use the ‘Request for Information’ to get FDA’ assessment whether your product is a medical device and, if so, which generic type of device and class applies.
Although the authority is not required to answer the first question (medical device yes or no) as part of the request, it usually does inform if the product seems or does not seem to qualify as a medical device. Otherwise, you would use the Request for Designation process.
2.2 Approval procedure
Since the classification is often linked to the question of whether there is a “Substantially Equivalent Device”, the FDA provides information related to the applicable approval pathway (PMA, 510(k), or neither) in a Request for Information.
However, the authority does not want to give its assessment of whether there is a sufficiently similar medical device (predicate device) as part of a Request for Information. A pre-submission meeting would be the appropriate choice to discuss this with FDA.
2.3 Further regulatory requirements
In addition to the approval procedure, the authority also provides information on other regulatory requirements which are applicable – including, where appropriate, an ‘enforcement discretion’. This is the authority’s waiver to demand regulatory compliance.
3. How to submit a Request for Information
The FDA describes how manufacturers must proceed in the guidance document FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act.
3.1 Step 1: Get informed
Of course, the authority would like you to search the relevant databases first before submitting a Request for Information:
It would help if you also read the relevant publications:
And finally, you may also contact FDA for specific matters:
- Small manufacturers: Division of Small Manufacturers International and Consumer Assistance: 800-638-2041 or 301-796-7100 or [email protected]
- Combination products: Office of Combination Products, 301-796-8930 or [email protected]
3.2 Step 2: Submit a Request for Information
Meanwhile, it is possible to submit the Request for Information electronically via eSTAR. You can download the appropriate template from the FDA’s eSTAR website.
Alternatively, manufacturers can submit using the eCopy format or by post in the traditional way. However, we recommend that you use the eSTAR format.
Your documents should include the following information:
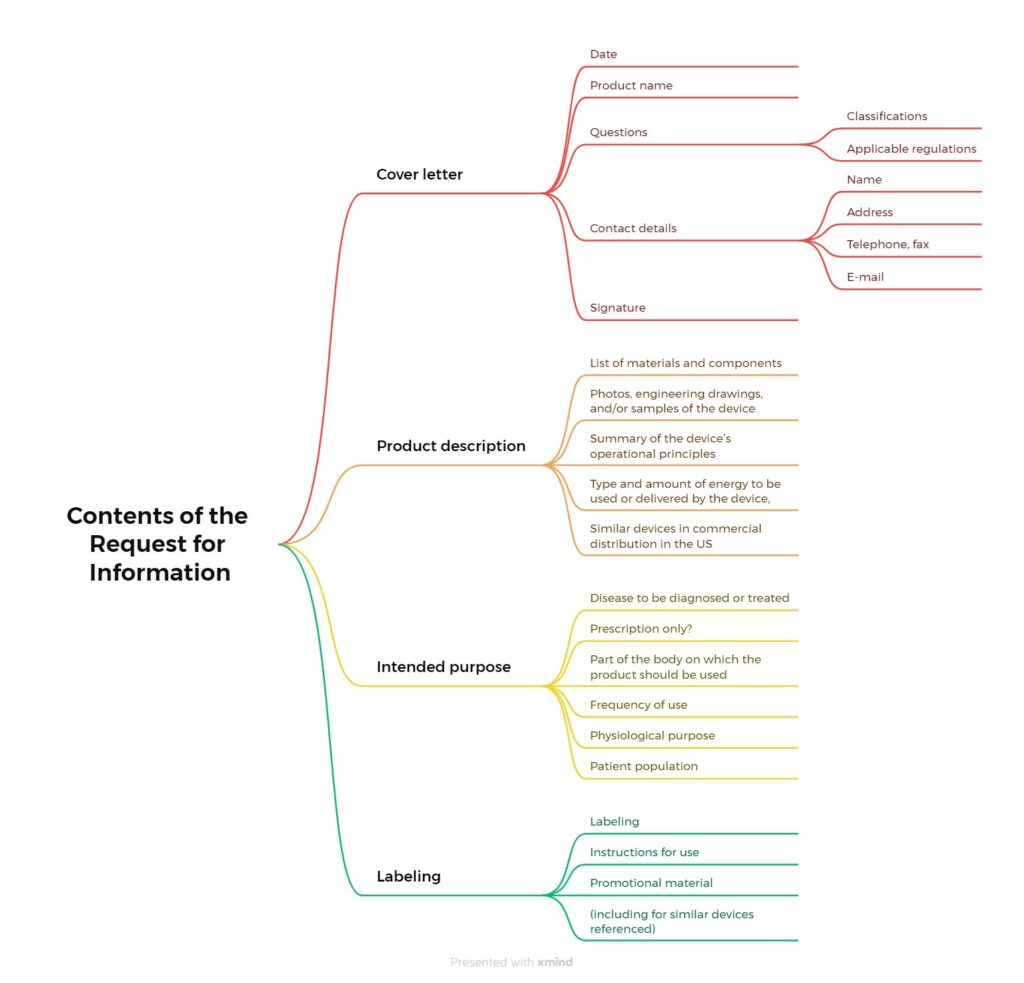
You must enclose a “Medical Device User Fee Cover Sheet” with the Request for Information. Instructions on how to complete this can be found here.
3.3 Step 3: Pay the fees
The FDA does not work for free either. The guidance document User Fees for 513(g) Requests for Information provides information on the fees. The fees exceed 7,000 USD, depending on the size of the company’. As a “small business”, you pay half of the actual fees.
Once you have submitted the application and paid the fees, there is no chance of a refund – not even if you withdraw the application. You also cannot supplement the application with further questions.
3.4 Step 4: Answer questions from the authority
FDA may ask for additional information, which you should provide promptly. There is no additional fee.
3.5 Step 5: Receive response from the authority
The FDA will respond in written form, usually within 60 days. The response letter is typically quite brief and includes:
- Generic type of device, if available
- Classification (I, II, III, “unclassified”, “not classified”)
- Approval procedure (exempt, 510(k), PMA)
- Other applicable regulatory requirements
- Contact information of other departments or offices of FDA, if it appears to be another type of product (e.g. combination product, other product type regulated by FDA or not a product type for which FDA has jurisdiction)
4. If you have any further questions
The FDA does not answer specific questions about the market approval of your device in the context of the ‘Request for Information’. Consider a ‘Pre-Submission’ in this case. You can find further information about pre-subs in this article.
Do you want to know how your device is classified in the US or do you need support with the Request for Information? Then get in touch with us right away!
Change history:
- 2024-11-18: Revision based on the updated guidance document. Reference to the eSTAR format.
