When asked what a design review is, you often get different answers – depending on whether you ask a developer or a quality manager. It is precisely these different views that can become a problem in the audit.
Definition: What is a design review?
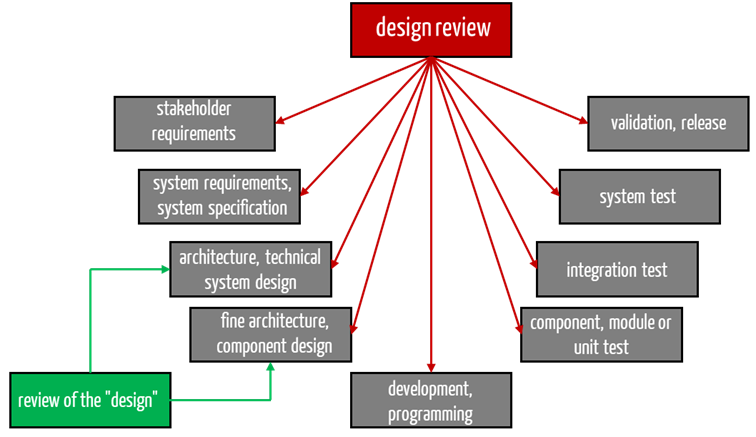
Many developers “misunderstand” the term design review to mean a review of the design. In the case of stand-alone software, the latter would correspond to what IEC 62304 means by verification of the architecture or detailed design. However, this does not correspond to the design review defined by ISO 13485 and the FDA (21 CFR part 820.30(e)).
Both regulations use the design review to mean the evaluation of the development. This evaluation should take place several times during or after development.
Objectives of the design review
The objective of the design review is to pause and check whether you are still on the right track. This concerns questions such as:
- Have we identified the right requirements of our customers?
- Are the (previous) design and development outputs suitable for meeting the requirements?
- Are there any new findings that those involved should be aware of?
- Is the device developed in such a way that it can later be produced without any problems?
- Have the risks in the sense of ISO 14971 been consistently analyzed, are they known to those involved, and have suitable measures been found and implemented?
- Is the project on track? Is it on schedule?
- Have all the documents that should be available for this project progress been created?
It is, therefore, about much more than just evaluating a system or software architecture. Rather, this design review should synchronize the team and critically scrutinize the entire development process.
Would you like support establishing or improving an ISO 13485 and/or FDA-compliant QM system? Professor Johner and his team will be happy to support you! Get in touch with us! Contact us
Design review: Regulatory requirements
ISO 13485 requirements for “design evaluation”
In chapter 7.3.5 ISO 13485 defines “design and development review.” The standard requires these evaluations “at suitable stages” with the objective of evaluating the ability of the design and development results to meet the requirements and to identify problems and propose necessary actions.
FDA requirements for the design review
The FDA defines a “design review” as a documented, complete, and systematic examination of the design with the objective of determining whether the requirements for the design are adequate, whether the design actually meets those requirements, and to identify problems. The CFR part 820.30(e) formulates the requirements for the design review:
- Each manufacturer shall establish and maintain procedures to ensure that formal documented reviews of the design results are planned and conducted at appropriate stages of the device’s design development.
- The procedures shall ensure that participants at each design review include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed.
- The results of a design review, including identification of the design, the date, and the individual(s) performing the review, shall be documented in the design history file (the DHF).
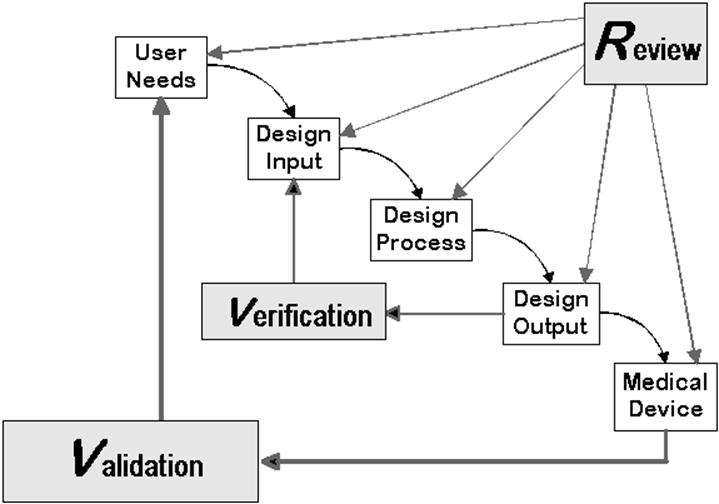
Comparison of FDA and ISO 13485 requirements
Both regulations set out the objectives of the design review:
- investigating whether the design meets the requirements for the device or its design
- identify problems
Furthermore, both require
- reviews (plural), i.e., several of these inspections,
- the participation of the necessary representatives
- to carry out the design reviews in accordance with a documented procedure and also to document the output.
Both regulations are, therefore, almost identical.
Planning and conducting a design review
A design review must be a documented procedure. Therefore, it must be defined in the QM system
- when the design review must take place. This can be, for example, at certain times or at the end of certain phases or milestones.
- which roles take part in this review. We recommend at least the development manager, quality manager, risk manager, product manager, and project manager.
- which aspects are to be examined during the review and
- how the output is to be documented.
If you need help to create an ISO 13485-compliant “Design Review” standard operating procedure (SOP), if you need templates or checklists, or if you would like support to establish your QM system completely and audit-proof within just a few days, please get in touch with us (web form).
Auditing a design review
The following aspects, for example, would be checked by an auditor as part of an internal audit or external audit:
- At what milestones or points in time does the manufacturer review the design?
- How does the focus of the design review, which takes place later in the development process, extend to the verification and validation of the medical devices?
- Can the manufacturer show what deviations or problems were encountered and what measures were taken?
- Which persons or roles took part in the reviews? Which specialists, if any, were consulted?
- Has the manufacturer documented the standard operating procedure, the output of the design reviews, and the measures taken? Do the outputs relate to a unique version of a device? Is the standard operating procedure controlled?
Our team also includes ISO 13485 lead auditors who will happily answer any questions you may have. Available free of charge.